36 Pan-Genome Analysis for Vaccine Development
- Learn how pan-genome analysis can inform vaccine development strategies.
- Explore two strategies for vaccine development using pan-genome data: targeting the core genome for broad-spectrum vaccines and targeting the accessory genome for virulence-specific vaccines.
- Gain familiarity with bioinformatics tools used in pan-genome analysis and genome-wide association studies (GWAS) for bacterial pathogens.
36.1 Pan-Genome Analysis for Vaccine Development
While reverse vaccinology is powerful for a single genome, its true potential is unlocked when applied to the pan-genome of a bacterial species. The pan-genome represents the entire set of genes found across all strains of a given species. It provides a complete picture of the genetic diversity and is crucial for designing vaccines with broad coverage.
As a reminder, the pan-genome is composed of three main parts:
Core Genome: Genes shared by all strains. These are typically essential for basic survival and are excellent targets for a broad-spectrum vaccine that aims to protect against the entire species.
Accessory Genome (or Dispensable Genome): Genes present in some, but not all, strains. This part of the genome often contains virulence factors, antibiotic resistance genes, and genes for adapting to specific environments. These are ideal for a targeted vaccine aimed at preventing disease caused by particularly virulent lineages.
Unique Genes: Genes specific to a single strain.
This approach allows for two distinct vaccine development strategies.
36.1.1 Strategy 1: Targeting the Core Genome for a Broad-Spectrum Vaccine
The goal here is to identify conserved proteins that are present in every strain of the pathogen, ensuring that the vaccine will be effective regardless of which strain a person is infected with.
Core genome workflow
Genome Collection and Annotation:
Input: Dozens to hundreds of high-quality genome sequences from diverse strains of the target bacterial species.
Process: Each genome is annotated (using a tool like Bakta) to identify all its protein-coding genes.
Pan-Genome Analysis:
Tool: A specialized pan-genome tool like Roary or Panaroo is used.
Process: The software compares all the annotated genes from every genome and clusters them into orthologous groups (gene families). It then categorizes each gene family as “core” (present in >99% of strains) or “accessory.”
Core Genome Candidate Selection (In Silico):
The list of core genes is filtered using the same reverse vaccinology criteria:
Predicted Location: The protein must be on the Outer Membrane or Secreted to be accessible to the immune system (predicted with PSORTb or SignalP).
Safety Screen: The protein must have no significant homology to human proteins to avoid autoimmunity (checked with DIAMOND or BLASTp against the human proteome).
Virulence Potential: Proteins with functions related to adhesion, invasion, or nutrient acquisition are prioritized.
Downstream Validation:
- The handful of promising core proteins are then produced in the lab and tested immunologically, following the standard reverse vaccinology pipeline.
Pan-Genome Analysis
We can use Panaroo to calculate the pan-genome of our set of Neisseria meningitidis genomes and identify the core genes. We can then parse the Panaroo output to extract the amino acid sequences of the core genes which can be used as input for the reverse vaccinology workflow described in the previous chapter.
Reverse Vaccinology Workflow on Core Genes
Now that we have identified the core genes using Panaroo, we can proceed with the reverse vaccinology workflow as described in the previous section. The core gene sequences extracted from Panaroo will serve as the input for subcellular localization prediction, homology searches, and functional annotation to identify potential vaccine candidates.
36.1.2 Strategy 2: Targeting the Accessory Genome for a Virulence-Specific Vaccine
This strategy focuses on identifying genes that are not essential for the bacterium’s survival but are strongly associated with its ability to cause severe disease. A vaccine targeting these proteins would not necessarily prevent colonization but would prevent the development of invasive disease.
Accessory Genome Workflow
Genome Collection with Metadata:
- Input: This requires a well-curated set of genomes where each strain is labeled with clinical metadata (e.g., “invasive disease” vs. “asymptomatic carriage”).
Pan-Genome Analysis:
- The process is the same as above, using panaroo or a similar tool to generate a complete pan-genome and classify genes as core or accessory.
Genome-Wide Association Study (GWAS):
Tool: A bacterial GWAS tool like Scoary or Pyseer is used.
Process: The tool takes the pan-genome output (a presence/absence matrix of all accessory genes) and the clinical metadata. It then performs statistical tests to find which accessory genes are significantly more common in the “invasive disease” group compared to the “carriage” group.
Virulent Gene Candidate Selection:
- The list of disease-associated accessory genes is then filtered using the standard reverse vaccinology criteria (subcellular location, human homology).
Downstream Validation:
- The top candidates are validated in the lab. A successful vaccine would generate antibodies that neutralize these specific virulence factors, effectively disarming the pathogen.
Genome-Wide Association Study (GWAS) on Panaroo output
We can use pyseer to perform a GWAS on the accessory genome of our Neisseria meningitidis genomes to identify genes associated with invasive disease. We will use the gene presence/absence matrix generated by Panaroo and a phenotype file indicating which strains are associated with invasive disease.
The first step is to estimate the population structure of our genomes (we do this to filter out the noise from shared ancestry and find the specific genes that are truly linked to our trait). We will do this using a pairwise distance matrix produced using mash:
# activate the accessory-vaccinology environment
mamba activate accessory-vaccinology
# create output directory for mash results
mkdir -p results/mash
# create mash sketch from genome assemblies
mash sketch -s 10000 -o results/mash/mash_sketch data/genomes/*.fa
# compute pairwise distances
mash dist results/mash/mash_sketch.msh results/mash/mash_sketch.msh | square_mash > results/mash/mash.tsv
# clean up the strain names in the mash distance file
sed -i 's/_contigs//g' results/mash/mash.tsvLet’s perform an MDS (multi-dimensional scaling) on these distances and look at a scree plot to choose the number of dimensions (a measure of our population structure) to retain:
scree_plot_pyseer results/mash/mash.tsv --output results/mash/scree_plot.pngWe got the following scree plot:
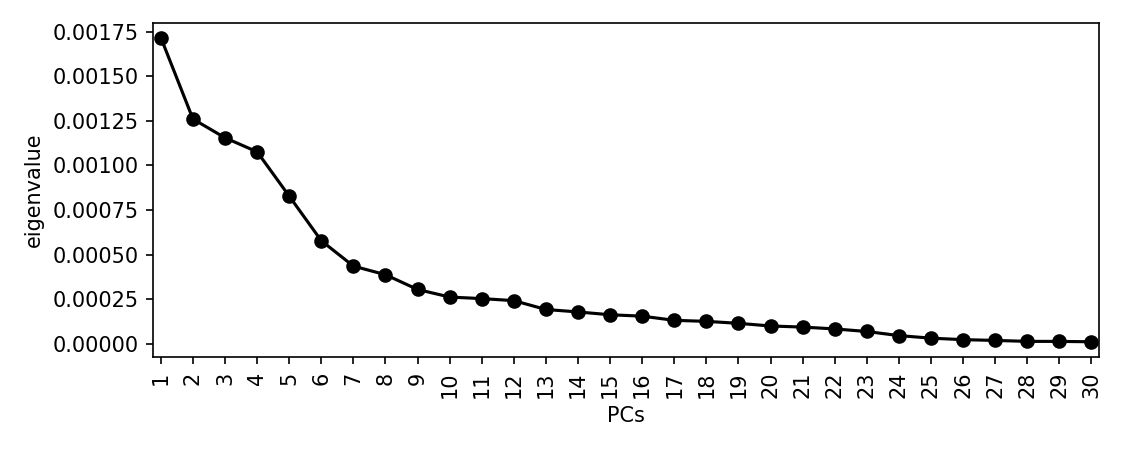
There is a drop after about 9 dimensions, so we will use this many. This is subjective, and you may choose to include many more. This is a sensitivity/specificity tradeoff – choosing more components is more likely to reduce false positives from population structure, at the expense of power. Using more components will also slightly increase computation time.
We can now run the analysis on the gene_presence_absence.Rtab file produced by Panaroo, using the phenotype file resources/gwas/disease.pheno, which indicates which strains are associated with invasive disease:
# create output directory for pyseer results
mkdir -p results/pyseer
pyseer --phenotypes resources/gwas/disease.pheno \
--pres results/panaroo/gene_presence_absence.Rtab \
--distances results/mash/mash.tsv \
--save-m results/pyseer/mash_mds \
--max-dimensions 9 \
> results/pyseer/disease_COGs.txtThe options we used are:
--phenotypes: the phenotype file indicating which strains are associated with invasive disease.--pres: the gene presence/absence matrix from Panaroo.--distances: the pairwise distance matrix from mash.--save-m: save the MDS components to a file for later use.--max-dimensions: the number of MDS dimensions to include as covariates in the model.
The output file results/pyseer/disease_COGs.txt contains the results of the pyseer GWAS analysis. We can filter this file to identify genes that are significantly associated with invasive disease (e.g., using a p-value threshold of 0.05 after Bonferroni correction). We can use an awk command to filter the results:
awk -F'\t' 'NR == 1 || ($4 < 0.05 && $17 !~ /bad-chisq/ && $17 !~ /high-bse/)' results/pyseer/disease_COGs.txt > results/pyseer/significant_hits.tsvThe resulting file results/pyseer/significant_hits.tsv contains the 9 genes that are significantly associated with invasive disease, which can be further analyzed to identify potential vaccine candidates:
variant af filter-pvalue lrt-pvalue
group_2118 8.09E-01 4.94E-01 4.26E-02
group_1010 5.88E-01 1.33E-01 3.63E-02
group_1113 5.29E-01 7.62E-01 4.06E-02
group_762 5.15E-01 4.80E-01 3.58E-02
group_185 4.56E-01 5.86E-02 2.54E-03
group_1006 4.12E-01 3.56E-01 1.90E-02
group_266 3.53E-01 1.36E-03 4.93E-02
group_983 3.38E-01 1.45E-01 2.47E-02
group_2141 3.09E-01 5.12E-01 2.97E-0236.2 Summary
- Pan-genome analysis enhances reverse vaccinology by considering the full genetic diversity of a bacterial species.
- Targeting the core genome allows for the development of broad-spectrum vaccines effective against all strains.
- Targeting the accessory genome enables the design of vaccines that specifically prevent invasive disease by neutralizing virulence factors.
- Bioinformatics tools like Roary, Panaroo, Scoary, and Pyseer are essential for pan-genome analysis and GWAS in bacterial vaccine development.