7 The nf-core project
- Describe what nf-core is and how it relates to Nextflow.
- Search for public nf-core pipelines and access their help documentation.
- Understand how to configure nf-core pipelines.
7.1 What is nf-core?
nf-core is a community-led project to develop a set of best-practice pipelines built using Nextflow workflow management system. Pipelines are governed by a set of guidelines, enforced by community code reviews and automatic code testing.

7.2 What are nf-core pipelines?
nf-core pipelines are an organised collection of Nextflow scripts, other non-nextflow scripts (written in any language), configuration files, software specifications, and documentation hosted on GitHub. There is generally a single pipeline for a given data and analysis type, e.g. there is a single pipeline for bulk RNA-Seq. All nf-core pipelines are distributed under the open MIT licence.
7.3 Running nf-core pipelines
7.3.1 Software requirements for nf-core pipelines
nf-core pipeline software dependencies are specified using either Docker, Singularity or Conda. It is Nextflow that handles the downloading of containers and creation of conda environments. In theory it is possible to run the pipelines with software installed by other methods (e.g. environment modules, or manual installation), but this is not recommended.
7.4 Usage instructions and documentation
You can find general documentation and instructions for Nextflow and nf-core on the nf-core website . Pipeline-specific documentation is bundled with each pipeline in the /docs folder. This can be read either locally, on GitHub, or on the nf-core website.
Each pipeline has its own webpage e.g. nf-co.re/rnaseq.
In addition to this documentation, each pipeline comes with basic command line reference. This can be seen by running the pipeline with the parameter --help . It is also recommended to explicitly specify the version of the pipeline you want to run, to ensure reproducibility if you run it again in the future. This can be done with the -r option.
For example, the following command prints the help documentation for version 3.4 of the nf-core/rnaseq pipeline:
nextflow run -r 3.4 nf-core/rnaseq --helpN E X T F L O W ~ version 20.10.0
Launching `nf-core/rnaseq` [silly_miescher] - revision: 964425e3fd [3.4]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/rnaseq v3.0
------------------------------------------------------
Typical pipeline command:
nextflow run nf-core/rnaseq --input samplesheet.csv --genome GRCh37 -profile docker
Input/output options
--input [string] Path to comma-separated file containing information about the samples in the experiment.
--outdir [string] Path to the output directory where the results will be saved.
--public_data_ids [string] File containing SRA/ENA/GEO identifiers one per line in order to download their associated FastQ files.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
--skip_sra_fastq_download [boolean] Only download metadata for public data database ids and don't download the FastQ files.
--save_merged_fastq [boolean] Save FastQ files after merging re-sequenced libraries in the results directory.
..truncated..7.4.1 Config files
nf-core pipelines make use of Nextflow’s configuration files to specify how the pipelines runs, define custom parameters and what software management system to use e.g. docker, singularity or conda.
Nextflow can load pipeline configurations from multiple locations. nf-core pipelines load configuration in the following order:
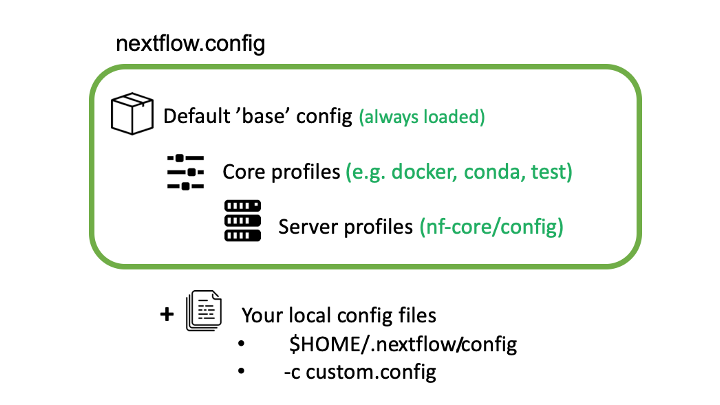
Pipeline: Default ‘base’ config
- Always loaded. Contains pipeline-specific parameters and “sensible defaults” for things like computational requirements.
- Does not specify any method for software packaging. If nothing else is specified, Nextflow will expect all software to be available on the command line.
Core config profiles
- All nf-core pipelines come with some generic config profiles. The most commonly used ones are for software packaging: docker, singularity and conda.
- Other core profiles are ‘debug’ and two ‘test’ profiles. The two test profiles include: a small version for quick testing, which pulls data from a public repository at nf-core/test-datasets; and a full test profile which provides the path to full-sized data from public repositories.
Server profiles
- At run time, nf-core pipelines fetch configuration profiles from the configs remote repository. The profiles here are specific to clusters at different institutions.
- Because this is loaded at run time, anyone can add a profile here for their system and it will be immediately available for all nf-core pipelines.
Local config files given to Nextflow with the
-cflagnextflow run nf-core/rnaseq -r 3.4 -c mylocal.configCommand line configuration: pipeline parameters can be passed on the command line using the
--<parameter>syntax. We will see several examples of this use throughout the course.
7.4.2 Config profiles
nf-core makes use of Nextflow configuration profiles to make it easy to apply a group of options on the command line.
Configuration files can contain the definition of one or more profiles. A profile is a set of configuration attributes that can be activated/chosen when launching a pipeline execution by using the -profile command line option. Common profiles are conda, singularity and docker that specify which software manager to use.
Multiple profiles are comma-separated. When there are differing configuration settings provided by different profiles, the right-most profile takes priority.
For example, the following command runs the test profile (i.e. run the test pipeline) and use Singularity containers as a way to manage the software required by the pipeline:
nextflow run nf-core/rnaseq -r 3.4 -profile test,singularityNote The order in which config profiles are specified matters. Their priority increases from left to right.
Be clever with multiple Nextflow configuration locations. For example, use -profile for your cluster configuration, the file $HOME/.nextflow/config for your personal config such as params.email and a working directory nextflow.config file for reproducible run-specific configuration.
7.4.3 Running pipelines with test data
The nf-core config profile test is a special profile, which defines a minimal data set and configuration, that runs quickly and tests the workflow from beginning to end. Since the data is minimal, the output is often nonsense. Real world example output are instead linked on the nf-core pipeline web page, where the workflow has been run with a full size data set:
$ nextflow run nf-core/<pipeline_name> -r "<pipeline_version>" -profile testNote that you will typically still need to combine this with a software configuration profile for your system - e.g. -profile test,conda. Running with the test profile is a great way to confirm that you have Nextflow configured properly for your system before attempting to run with real data
7.5 Troubleshooting
If you run into issues running your pipeline you can you the nf-core website to troubleshoot common mistakes and issues https://nf-co.re/usage/troubleshooting .
7.5.1 Extra resources and getting help
If you still have an issue with running the pipeline then feel free to contact the nf-core community via the Slack channel . The nf-core Slack workspace has channels dedicated for each pipeline, as well as specific topics (eg. #help, #pipelines, #tools, #configs and much more). To join this workspace you will need an invite, which you can get at https://nf-co.re/join/slack.
You can also get help by opening an issue in the respective pipeline repository on GitHub asking for help.
If you have problems that are directly related to Nextflow and not our pipelines or the nf-core framework tools then check out the Nextflow gitter channel or the google group.
7.6 Referencing a Pipeline
7.6.1 Publications
If you use an nf-core pipeline in your work you should cite the main publication for the main nf-core paper, describing the community and framework, as follows:
The nf-core framework for community-curated bioinformatics pipelines. Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen. Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x. ReadCube: Full Access Link
Many of the pipelines have a publication listed on the nf-core website that can be found here.
7.6.2 DOIs
In addition, each release of an nf-core pipeline has a digital object identifiers (DOIs) for easy referencing in literature The DOIs are generated by Zenodo from the pipeline’s github repository.
7.7 Summary
- nf-core is a community-led project to develop and curate a set of high-quality bioinformatic pipelines.
- All pipelines are listed on the nf-co.re website.
- Each pipeline contains both general usage documentation, as well as detailed help for its input parameters and the output files generated by the pipeline.
- Several aspects of the pipelines can be configured, using configuration files.
- Default profiles can be used to load a set of default configurations. Common profiles in nf-core pipelines include:
testto run a quick test, useful to see if the software is correctly setup on the computer/server being used.dockerandsingularityto indicate we want to use either Docker or Singularity to automatically manage the software installation during the pipeline run.
7.8 Credit
Information on this page has been adapted and modified from the following source(s):
- Graeme R. Grimes, Evan Floden, Paolo Di Tommaso, Phil Ewels and Maxime Garcia. Introduction to Workflows with Nextflow and nf-core. https://github.com/carpentries-incubator/workflows-nextflow 2021.