33 Pathogenwatch 2
TipLearning Objectives
- Analyse S. penumoniae genomes on Pathogenwatch and extract relevant information to use as metadata to annotate phylogenetic trees.
ExerciseExercise 1 - Analysing Pneumococcal genomes with Pathogenwatch
Whilst you can upload FASTQ files to Pathogenwatch, it’s quicker if we work with already assembled genomes. We’ve provided pre-processed results for the S. pneumoniae data generated with assembleBAC in the preprocessed/ directory which you can use to upload to Pathogenwatch in this exercise.
- Upload the assembled S. pneumoniae genomes to
Pathogenwatch. - Once
Pathogenwatchhas finished processing the genomes, save the results to a collection called Chaguza Serotype 1. - Download the Typing and AMR profile tables to the
S_pneumoniaedirectory. - Rename the tables to
chaguza-serotype-1-typing.csvandchaguza-serotype-1-amr-profile.csvrespectively. - Merge the two tables with
sample_info.csvby running themerge_pneumo_data.pyscript in thescriptsdirectory. Make sure you are on thebasesoftware environment.- Note: If you have not managed to run the Pathogenwatch analysis, we provide the output files from the previous steps in
preprocessed/pathogenwatchthat you can use instead.
- Note: If you have not managed to run the Pathogenwatch analysis, we provide the output files from the previous steps in
AnswerAnswer
- We opened
Pathogenwatchin our web browser and logged in. We then clicked on UPLOAD, the Upload FASTA(s) button in the “Single Genome FASTAs” section and the + button before navigating to thepreprocessed/assemblebac/assembliesdirectory. We then selected all the assembly files and clicked Open on the dialogue window. - We waited for
Pathogenwatchto finish processing the genomes, clicked the VIEW GENOMES button, then saved the results to a collection called Chaguza Serotype 1 by clicking Selected Genomes –> Create Collection and adding Chaguza Serotype 1 to the Title box. Finally, we clicked Create Now button to create our collection.
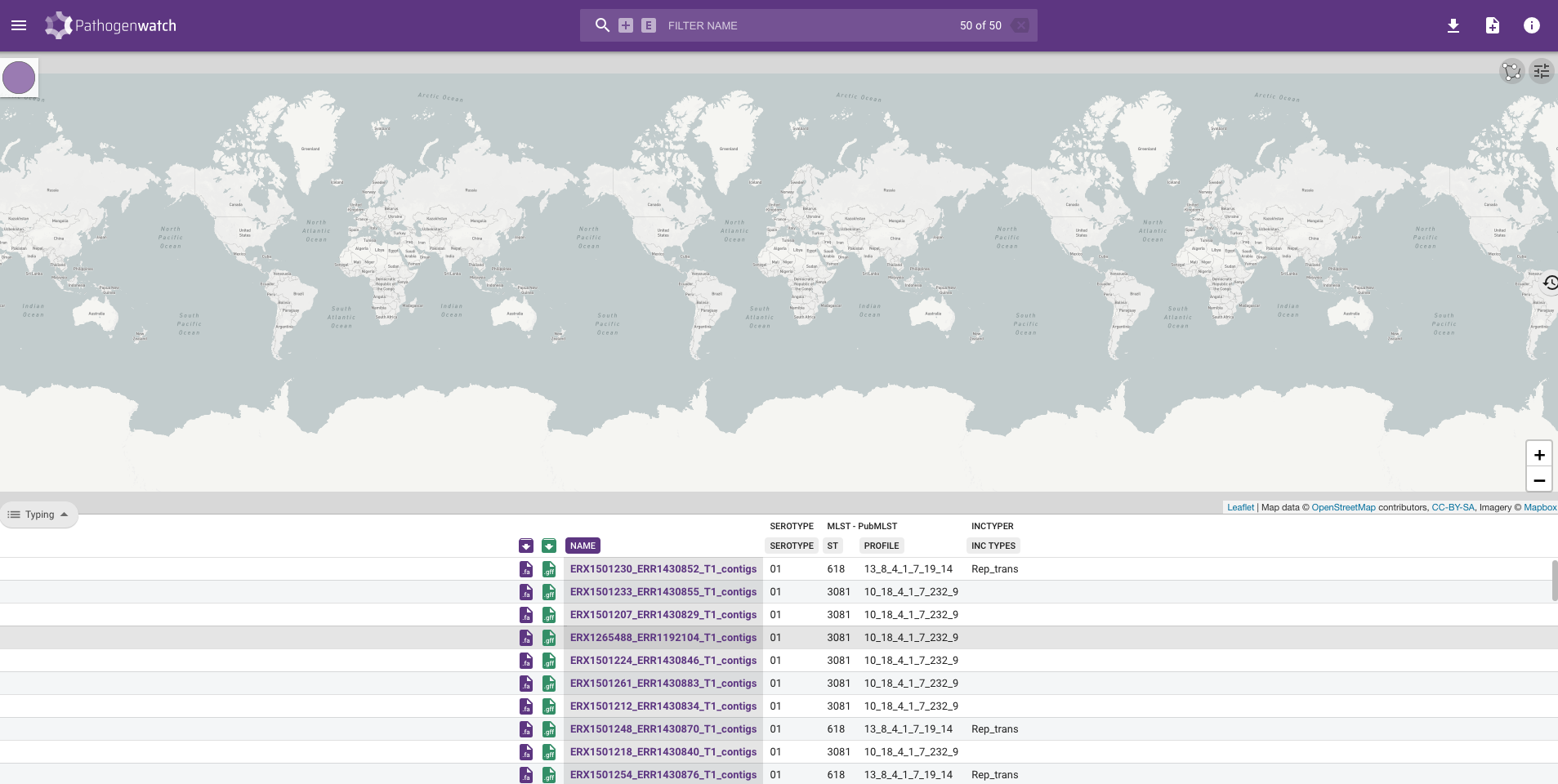
- We clicked on the download icon in the top right-hand corner and selected Typing table.
- We clicked on the download icon in the top right-hand corner and selected AMR profile.
- We renamed the files to
chaguza-serotype-1-typing.csvandchaguza-serotype-1-amr-profile.csvrespectively and moved them to theS_pneumoniaedirectory. - We went back to the
basesoftware environment withmamba activate base - We ran the
merge_pneumo_data.pyscript to create a TSV file calledpneumo_metadata.tsvin your analysis directory:
python scripts/merge_pneumo_data.py -s sample_info.csv -t chaguza-serotype-1-typing.csv -a chaguza-serotype-1-amr-profile.csv