15 TB-Profiler
- Describe how TB-profiler can be used to further annotate Mycobacterium tuberculosis genomes.
- Run TB-profiler on a single sample and on multiple samples using a bespoke script.
- Prepare TB-profiler data to annotate phylogenetic trees.
15.1 Introduction
TB-Profiler is a tool used to detect antimicrobial resistance and the lineages of MTBC genomes. It is made up of a pipeline which by default uses Trimmomatic to trim reads, aligns the reads to the H37Rv reference using bowtie2, BWA or minimap2 and then calls variants using bcftools. These variants are then compared to a drug-resistance database and a database of lineage-defining variants.
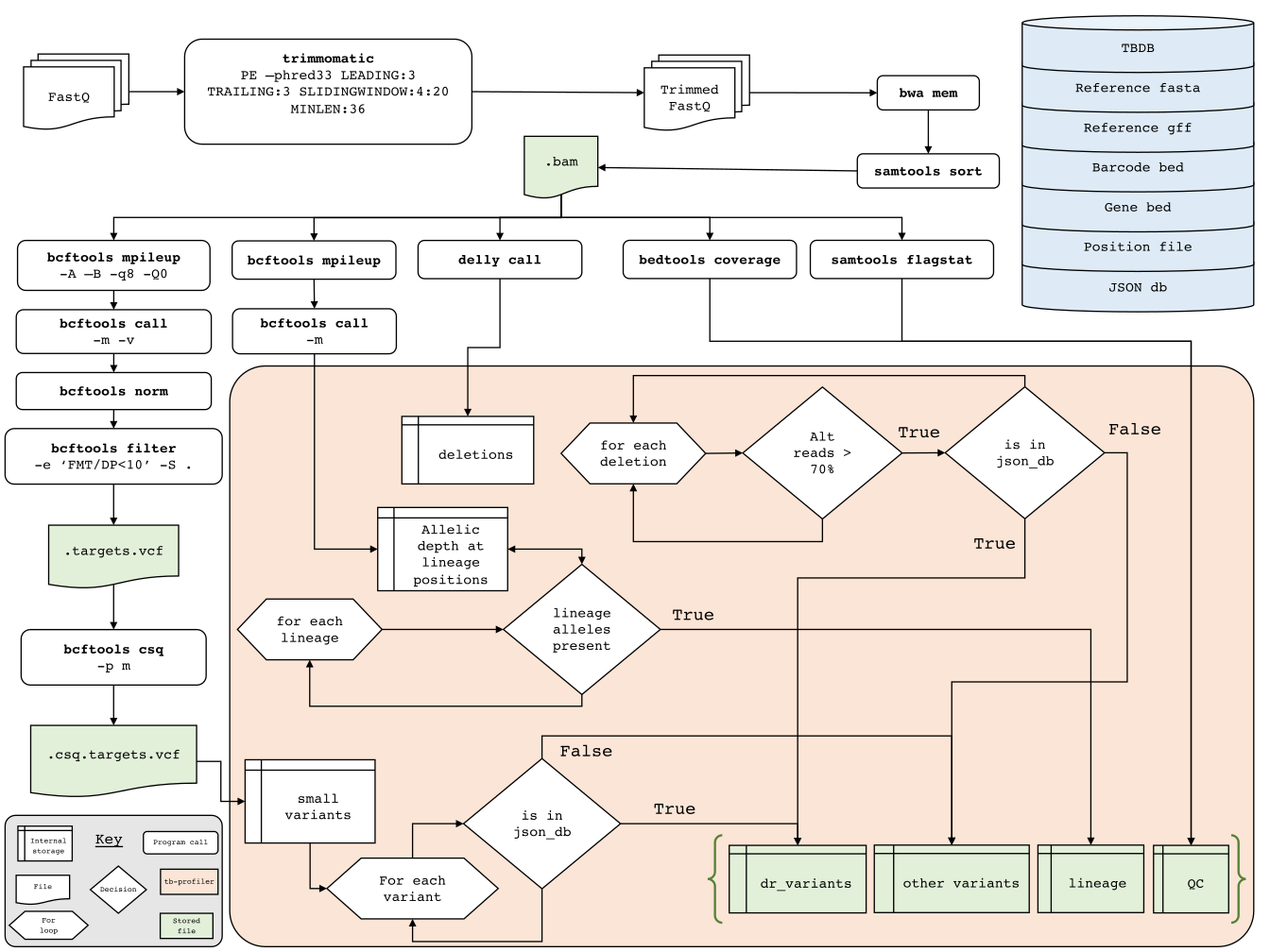
Note that, like many other database-based tools TB-Profiler is under constant rapid development. If you plan to use the program in your work please make sure you are using the most up to date version! Similarly, the database is not static and is continuously being improved so make sure you are using the most latest version. If you use TBProfiler in your work please state the version of both the tool and the database as they are developed independently from each other.
There is an online version of the tool which is very useful for analysing few genomes. You can try it out later at your free time by following this link.

15.2 Running TB-Profiler on the command line
Given we have five TB genomes to analyse, we’re going to run TB-Profiler on the command line instead of uploading the FASTQ files to the web version, starting with a single sample ERX9450498_ERR9907670.
We’ll start by activating the software environment:
mamba activate tb-profilerTo run TB-Profiler on ERX9450498_ERR9907670, the following commands can be used:
# create output directory
mkdir -p results/tb-profiler
# run TB-Profiler
tb-profiler profile -1 data/reads/ERX9450498_ERR9907670_1.fastq.gz -2 data/reads/ERX9450498_ERR9907670_2.fastq.gz -p ERX9450498_ERR9907670 -t 8 --csv -d results/tb-profiler 2> results/tb-profiler/ERX9450498_ERR9907670.logThe options we used are:
-1- the read 1 FASTQ file.-2- the read 2 FASTQ file.-p- the prefix of the output files, in this case the sample ID.-t- the number of CPUs to use.--csv- saves the output files in CSV format.-d- the name of the directory to save the results to.2>- to help with identifying if a sample fails we redirect any error messages to a log file.
While it was running it printed a message on the screen:
[17:24:20] INFO Using ref file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.fasta db.py:795
INFO Using gff file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.gff db.py:795
INFO Using bed file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.bed db.py:795
INFO Using version file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.version.json db.py:795
INFO Using json_db file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.dr.json db.py:795
INFO Using variables file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.variables.json db.py:795
INFO Using spoligotype_spacers file: db.py:795
/home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.spoligotype_spacers.txt
INFO Using spoligotype_annotations file: db.py:795
/home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.spoligotype_list.csv
INFO Using bedmask file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.mask.bed db.py:795
INFO Using barcode file: /home/ajv37/miniconda3/envs/tb-profiler/share/tbprofiler//tbdb.barcode.bed db.py:795
INFO Trimming readsYou can open the results file (results/tb-profiler/results/ERX9450498_ERR9907670.results.csv) with a spreadsheet software such as Excel from your file browser . There is a lot of information in this file but TB-Profiler helpfully provides a summary of the most useful information at the top:
TBProfiler report
=================
The following report has been generated by TBProfiler.
Summary
-------
ID ERX9450498_ERR9907670
Date Thu Nov 9 11:09:46 2023
Strain lineage4.3.4.1
Drug-resistance MDR-TB
Median Depth 84These key fields are:
- ID - our sample ID.
- Date - the date and time TB-Profiler was run on this sample.
- Strain - the lineage of the sample.
- Drug-resistance - the drug resistance profile of the sample.
- Median Depth - the median read depth coverage across the reference genome.
From these results we can see that sample ERX9450498_ERR9907670 belongs to lineage 4.3.4.1 and is multi-drug resistant (MDR) i.e. resistant to more than one drug, in this case Rifampicin, Isoniazid and Ethambutol.
15.3 TB-Profiler collate
TB-Profiler has an option to collect the outputs from multiple samples and collate these together into a useful summary CSV file. We’re going to use this command in the exercise below.
Hopefully, we’ll be able to successfully run TB-Profiler on all our samples but, unfortunately, sometimes it’ll fail. If this does happen, identify which sample didn’t complete and re-run TB-Profiler. This is the reason we save the error messages to a log file.
15.4 Data cleaning
The final thing we need to do before we visualize our TB phylogeny is to combine the metadata contained in sample_info.csv with the TB-profiler so we have some interesting information to annotate our tree with. We provide a python script to do this for you but you could, of course, do this yourself in Excel or using R. You can run the script using the following command (make sure to mamba activate tb-profiler):
python scripts/merge_tb_data.py -s sample_info.csv -t preprocessed/tb-profiler/Nam_TB.txtThe options we used are:
-s- the CSV file containing the metadata extracted from the publication.-t- the TB-Profiler summary TSV file (in this case the version in thepreprocesseddirectory).
This will create a TSV file called TB_metadata.tsv in your analysis directory. We’ll use this file along with the TB phylogenetic tree in the next section.
15.5 Summary
- TB-profiler can be used to detect the presence of antimicrobial resistance genes in a genome, as well as assign it to its most likely lineage.
- TB-profiler can be run from the command line to process a single sample. Multiple samples can be conveniently processed using a for loop.
- TB-profiler outputs a CSV file with information about lineages and drug resistance genes.
- Multiple output files from TB-profiler can be combined with metadata of our samples to generate a TSV file to annotate phylogenetic trees.