31 Introduction to recombination
- Describe what recombination in bacteria is.
- Apply the Gubbins software to generate recombination-free alignments.
- Build a phylogeny from a recombination-free alignment.
31.1 Removing recombination
Recombination in bacteria is characterized by DNA transfer from one organism or strain (the donor) to another organism/strain (the recipient) or the uptake of exogenous DNA from the surrounding environment. Broadly, there are three different types of bacterial recombination:
- Transformation: the uptake of exogenous DNA from the environment
- Transduction: virus-mediated (phage) transfer of DNA between bacteria
- Conjugation: the transfer of DNA from one bacterium to another via cell-to-cell contact
The sequences transferred via recombination can influence genome-wide measures of sequence simularity more than vertically-inherited point mutations that are the signal of shared common ancestor. Thus, identifying recombinant regions and accounting for their potentially different phylogenetic history is crucial when examining the evolutionary history of bacteria. In practice, what this means is that we remove (mask) previously identified recombinant regions in our multiple sequence alignments, before we proceed with phylogenetic tree inference.
The two most commonly used tools to do this are Gubbins and ClonalFrameML. It’s important to note that Gubbins cannot be used on the core gene alignment produced by tools like roary or panaroo. Instead, Gubbins requires a whole-genome alignment as input, in order to analyse the spatial distribution of base substitutions. For this reason, the finer-scale phylogenetic structure of trees generated using a core gene alignment may be less accurate. If we want to properly account for recombination in this instance, typically we would perform clustering on our initial tree, then map sequence data for the samples within a cluster to a suitable reference before running our recombination removal tool of choice.
31.2 Gubbins
Gubbins (Genealogies Unbiased By recomBinations In Nucleotide Sequences) is an algorithm that iteratively identifies loci containing elevated densities of base substitutions, while concurrently constructing a phylogeny based on the putative point mutations outside of these regions. We’re going to use Gubbins to identify the recombinant regions in the alignment we generated using bactmap.
31.3 Running Gubbins
We’ll start by activating the gubbins software environment:
mamba activate gubbinsTo run Gubbins on the aligned_pseudogenomes.fas file, the following commands can be used:
# create output directory
mkdir -p results/gubbins/
# run gubbins
run_gubbins.py --prefix sero1 --tree-builder iqtree results/bactmap/pseudogenomes/aligned_pseudogenomes.fas
# move gubbins outputs to results directory
mv sero1.* results/gubbins/The options we used are:
--prefix- prefix to use for theGubbinsoutput files.--tree-builder-Gubbinscan be run with different phylogenetic software includingIQ-TREE,FastTreeandRAxML.
As it runs, Gubbins prints several messages to the screen.
We moved the output files to results/gubbins/ where we can see all the output files it generated:
ls results/gubbinssero1.final_tree.tre sero1.per_branch_statistics.csv sero1.summary_of_snp_distribution.vcf
sero1.branch_base_reconstruction.embl sero1.log sero1.recombination_predictions.embl
sero1.filtered_polymorphic_sites.fasta sero1.node_labelled.final_tree.tre sero1.recombination_predictions.gff
sero1.filtered_polymorphic_sites.phylip 31.4 Masking recombinant regions
In a similar way to how we masked the TB alignment to remove certain regions of the reference genome from downstream analyses, the next step is to mask the recombinant regions in our aligned_pseudogenomes.fas file, so these do not influence our phylogenetic tree inference. Instead of using remove_blocks_from_aln.py, we will use mask_gubbins_aln.py (included with Gubbins):
mask_gubbins_aln.py --aln results/bactmap/pseudogenomes/aligned_pseudogenomes.fas --gff results/gubbins/sero1.recombination_predictions.gff --out results/gubbins/aligned_pseudogenomes_masked.fasThe options we used are:
--aln- the input alignment, in this case the alignment created bybactmap.--gff- the GFF file containing the coordinates for the recombinant regions identified byGubbins.--out- the masked alignment.
The masked final alignment will be saved to the results/gubbins/ directory.
31.5 Visualizing recombinant regions
The outputs from Gubbins can be visualised by running a R script included as part of Gubbins:
plot_gubbins.R -t results/gubbins/sero1.final_tree.tre -r results/gubbins/sero1.recombination_predictions.gff -a resources/reference/GCF_000299015.1_ASM29901v1_genomic.gff -o results/gubbins/sero1.recombination.pngThe options we used are:
-t- the recombination-free phylogenetic tree created byGubbins.-r- the GFF file containing the coordinates for the recombinant regions identified byGubbins.-a- the GFF file for the reference genome containing the coordinates for the coding regions.-o- the figure showing the recombinant regions identified byGubbins.
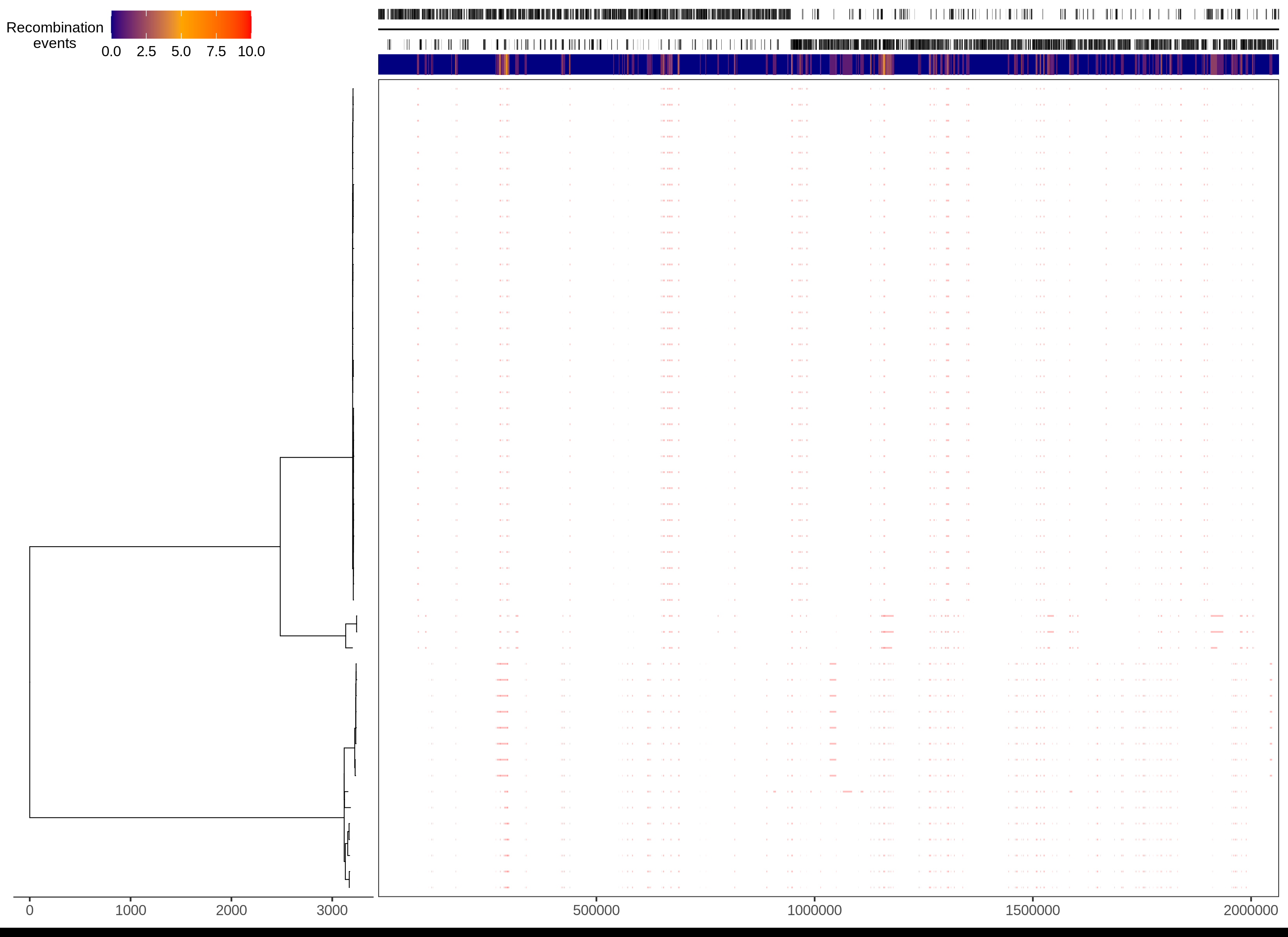
The panel on the left shows the maximum-likelihood phylogeny built from the clonal frame of serotype isolates. The scale below shows the length of branches in base substitutions. The tree is coloured according to the classification of isolates, each of which corresponds to a row in the panel on the right. Each column in this panel is a base in the reference annotation, the annotation of which is shown at the top of the figure. The panel shows the distribution of inferred recombination events, which are coloured blue if they are unique to a single isolate, or red, if they are shared by multiple isolates through common ancestry.
31.6 Summary
- In bacteria, recombination refers to the process of genetic material exchange between organisms, through processes such as transformation, transduction and conjugation.
- The Gubbins software can be to produce recombination-free alignments. It requires as input a multiple sequence alignment from whole genomes.
- After identifying recombinant regions, these are masked from the alignment (i.e. converted to ’N’s).
- The output from Gubbins can then be used as input to SNP-sites and IQ-tree as demonstrated before.