8 Downloading sequence data
- Download sequence data from a public database with the nf-core pipeline fetchngs.
8.1 Pipeline Overview
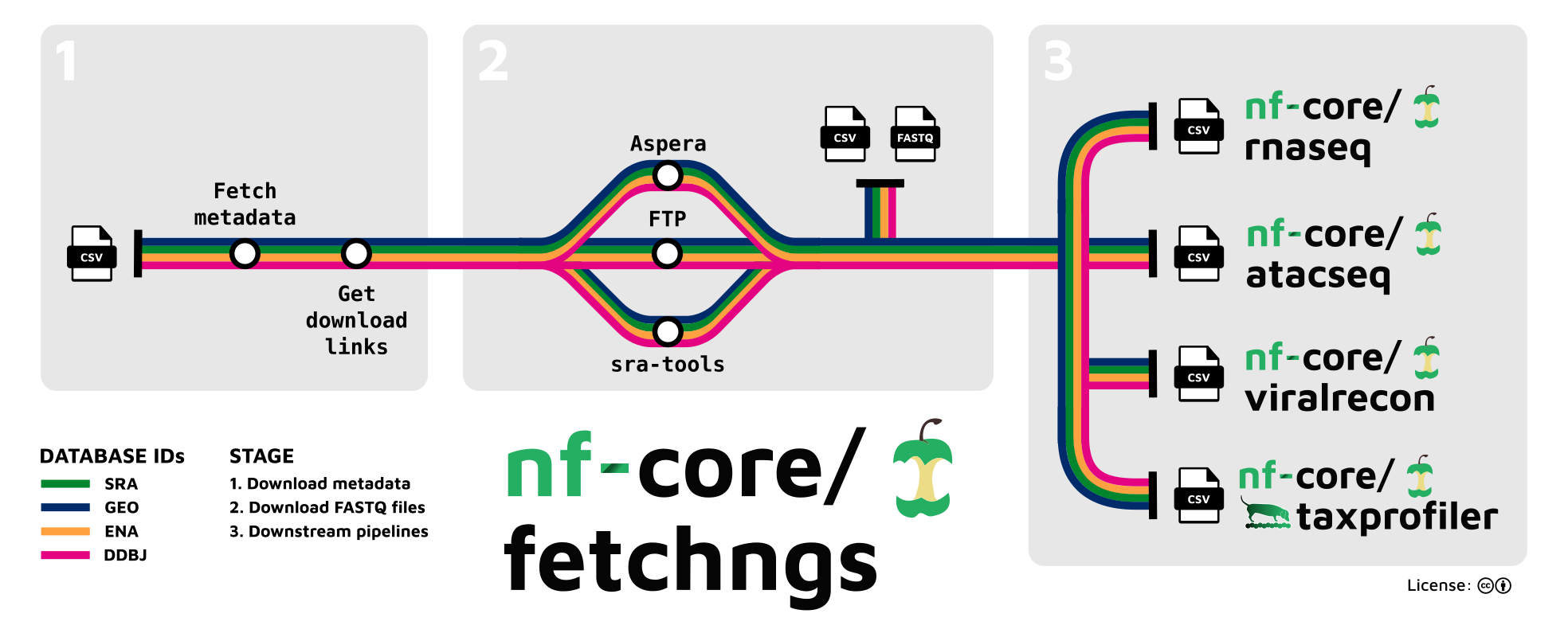
fetchngs is a bioinformatics analysis pipeline written in Nextflow to automatically download and process raw FASTQ files from public databases. Identifiers can be provided in a file and any type of accession ID found in the SRA, ENA, DDBJ and GEO databases are supported. If run accessions (SRR/ERR/DRR) are provided, these will be resolved back to the sample accessions (SRX/ERX/DRX) to allow multiple runs for the same sample to be merged. As well as the FASTQ files, fetchngs will also produce a samplesheet.csv file containing the sample metadata obtained from the ENA. This file can be used as input for other nf-core and Nextflow pipelines like the ones we’ll be using this week.
8.2 Prepare a samples file
fetchngs requires a samples file with the accessions you would like to download. The file requires the suffix .csv but does not need to be in CSV format. Each line needs to represent a database id:
ERR9907668
ERR9907669
ERR9907670
ERR9907671
ERR99076728.3 Running fetchngs
Now that we have the samples.csv file, we can run the fetchngs pipeline. First, let’s activate the nextflow software environment:
mamba activate nextflowThere are many options that can be used to customise the pipeline but a typical command is shown below:
nextflow run nf-core/fetchngs \
-r "1.12.0" \
-profile singularity \
--input SAMPLES \
--outdir results/fetchngs \
--nf_core_pipeline viralrecon \
--download_method sratools \
-resumeThe options we used are:
-profile singularity- indicates we want to use the Singularity program to manage all the software required by the pipeline (another option is to usedocker). See Data & Setup for details about their installation.--input- the samples file with the accessions to be downloaded, as explained above.--nf_core_pipeline- Name of supported nf-core pipeline e.g. ‘viralrecon’. A samplesheet for direct use with the pipeline will be created with the appropriate columns.--download_method- forces the pipeline to usesratoolsinstead of a direct FTP download.-resume- all Nextflow pipelines can be resumed. It isn’t necessary for the force run of the pipeline but it’s good practice to include it in the command.
8.4 fetchngs results
Once fetchngs has run, we can look at the various directories it created in results/fetchngs:
| Directory | Description |
|---|---|
custom |
Contains settings to help the pipeline run |
fastq |
Paired-end/single-end reads downloaded from the SRA/ENA/DDBJ/GEO for each accession in the samples.csv file |
metadata |
Contains the re-formatted ENA metadata for each sample |
samplesheet |
Contains the samplesheet with collated metadata and paths to downloaded FASTQ files |
pipeline_info |
Contains information about the pipeline run |
Each step of the pipeline produces one or more files that are not saved to the results directory but are kept in the work directory. This means that if, for whatever reason, the pipeline doesn’t finish successfully you can resume it. However, once the pipeline has completed successfully, you no longer need this directory (it can take up a lot of space) so you can delete it:
rm -rf work8.5 Summary
- The nf-core fetchngs pipeline can be used to quickly download sequence data from public databases such as the ENA and GEO.
- The pipeline also produces a
samplesheet.csvfile that can be used as input for other nf-core and Nextflow pipelines.