15 Managing Bioinformatics Software
- List some of the reasons why Linux is an essential operating system for bioinformatic analysis.
- Distinguish between software containers and software environments.
- Use package managers to install software globally and locally.
- Recognise the advantage of using software containers for bioinformatic pipelines.
15.1 Why Linux?
Most computer users either use Windows or macOS as their operating system. However, in bioinformatics the Linux operating system is the most popular.
There are different reasons to prefer Linux, for example:
- Most bioinformatics software is only available for this operating system.
- Generally more secure.
- Free and open source.
- The operating system of choice on high performance compute (HPC) cluster environments.
- It has flexible built-in command line tools to manipulate large data (
cat,grep,sed, and the|pipe for chaining commands).
Because Linux is an open source project, there are many different types of Linux operating systems available. Generally, we recommend using Ubuntu, as it’s a well-supported and widely used distribution.
The first step for running bioinformatics software therefore is to ensure that you have access to Ubuntu Linux on your machine. We give instructions for this in Section 16.1.
15.2 Package Managers
Once you have Linux available, there is the question of how to install software, especially when using the command line. Most modern Linux operating systems have a default package manager available. A package manager allows the user to install (or remove, or upgrade) software with a single command. The package manager takes care of automatically downloading and installing the software we want, as well as any dependencies it requires.
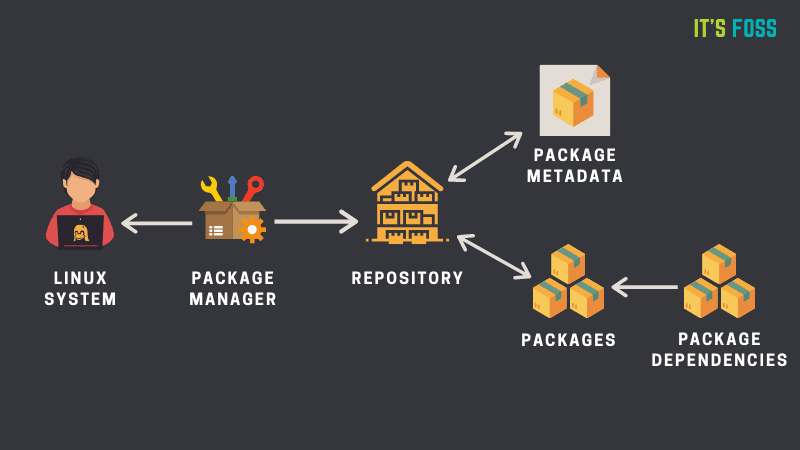
15.2.1 Ubuntu apt
On Ubuntu, the default package manager is called apt. This software can be used to do system-wide updates, upgrades and installation of software packages. There are several commands available:
apt updateupdates the database of available packages, to check the latest versions available. This command doesn’t actually install anything.apt upgradeupgrades all the packages to their latest version. This command upgrades existing packages, and installs new ones if required as dependencies.apt installis used to install a single software of your choice.
For example, let’s say we wanted to install the latest version of java (Java is used for many applications, including IGV, which we introduced earlier). We could run the following commands:
sudo apt update # make sure the package database is up-to-date
sudo apt upgrade # upgrade existing software to their latest version
sudo apt install default-jre # install javaThe sudo command at the beginning indicates that we want to run the apt command with administrator privilege, which will require you to input your password. This is necessary, because the apt command does a system installation of the software, so it can only be done if the user trying to do the installation has those permissions. With the install command, we give the name of the software we want to install. In this case, default-jre will install the default Java runtime environment.
The apt package manager is extremely useful to install core system software, however most bioinformatics software are not available from apt. Also, as it requires administrator (sudo) permissions, it is not always possible to install software with apt (for example, if you are using a HPC cluster). This is why we turn to alternative package managers such as Conda.
15.2.2 Debian Packages
Sometimes, software is not available through the apt repositories, but instead distributed as a file. Ubuntu uses the Debian package format. To install Debian packages, you can use the dpkg command.
For example, the RStudio application for Linux is distributed as a .deb file. After you download it, you can install it as follows:
sudo dpkg -i rstudio-2023.12.0-369-amd64.debAs with apt this is a system installation and so requires admin privileges (sudo).
15.2.3 Conda/Mamba Package Manager
Often you may want to use software packages that are not available on the apt repositories. A popular alternative in bioinformatics is to use the package manager Mamba, which is a successor to another package manager called Conda.
Conda and Mamba are package managers commonly used in data science, scientific computing, and bioinformatics. Conda, originally developed by Anaconda, is a package manager and environment manager that simplifies the creation, distribution, and management of software environments containing different packages and dependencies. It is known for its cross-platform compatibility and ease of use. Mamba is a more recent and high-performance alternative to Conda. While it maintains compatibility with Conda’s package and environment management capabilities, Mamba is designed for faster dependency resolution and installation, making it a better choice nowadays.
One of the strengths of using Mamba to manage your software is that you can have different versions of your software installed alongside each other, organised in environments. Organising software packages into environments is extremely useful, as it allows to have a reproducible set of software versions that you can use and reuse in your projects.
For example, imagine you are working on two projects with different software requirements:
- Project A: requires Python 3.7, NumPy 1.15, and scikit-learn 0.20.
- Project B: requires Python 3.9, the latest version of NumPy, and TensorFlow 2.0.
If you don’t use environments, you would need to install and maintain these packages globally on your system. This can lead to several issues:
- Version conflicts: different projects may require different versions of the same library. For example, Project A might not be compatible with the latest NumPy, while Project B needs it.
- Dependency chaos: as your projects grow, you might install numerous packages, and they could interfere with each other, causing unexpected errors or instability.
- Difficulty collaborating: sharing your code with colleagues or collaborators becomes complex because they may have different versions of packages installed, leading to compatibility issues.

Environments allow you to create isolated, self-contained environments for each project, addressing these issues:
- Isolation: you can create a separate environment for each project using tools like Conda/Mamba. This ensures that the dependencies for one project don’t affect another.
- Version control: you can specify the exact versions of libraries and packages required for each project within its environment. This eliminates version conflicts and ensures reproducibility.
- Ease of collaboration: sharing your code and environment file makes it easy for collaborators to replicate your environment and run your project without worrying about conflicts.
- Simplified maintenance: if you need to update a library for one project, it won’t impact others. You can manage environments separately, making maintenance more straightforward.
Another advantage of using Mamba is that the software is installed locally (by default in your home directory), without the need for admin (sudo) permissions.
You can search for available packages from the anaconda.org website. Packages are organised into “channels”, which represent communities that develop and maintain the installation “recipes” for each software. The most popular channels for bioinformatics and data analysis are “bioconda” and “conda-forge”.
There are three main commands to use with Mamba:
mamba create -n ENVIRONMENT-NAME: this command creates a new software environment, which can be named as you want. Usually people name their environments to either match the name of the main package they are installating there (e.g. an environment calledpangolinif it’s to install the Pangolin software). Or, if you are installing several packages in the same environment, then you can name it as a topic (e.g. an environment calledrnaseqif it contains several packages for RNA-seq data analysis).mamba install -n ENVIRONMENT-NAME NAME-OF-PACKAGE: this command installs the desired package in the specified environment.mamba activate ENVIRONMENT-NAME: this command “activates” the environment, which means the software installed there becomes available from the terminal.
Let’s see a concrete example. If we wanted to install packages for phylogenetic analysis, we could do:
# create an environment named "phylo"
mamba create -n phylo
# install some software in that environment
mamba install -n phylo iqtree mafftIf we run the command:
mamba env listWe will get a list of environments we created, and “phylo” should be listed there. If we want to use the software we installed in that environment, then we can activate it:
mamba activate phyloAnd usually this changes your terminal to have the word (phylo) at the start of your prompt.
15.3 Software Containers
Software containerization is a way to package software and its dependencies in a single file. A software container can be thought of as a very small virtual machine, with everything needed to run that software stored inside that file. For this reason, software containerization solutions, such as Docker and Singularity, are widely used in bioinformatics.
Software containers ensure reproducibility, allowing the same analysis to run on different systems. They can run on a local computer or on a high-performance computing cluster, producing the same result. The software within a container is isolated from other software, addressing the issue of incompatible dependencies between tools (similarly to Mamba environments).
As we already saw, analysis pipelines can be very complex, using many tools, each with their own dependencies. Therefore, worflow managers such as Nextflow use software containers to run their analysis (both Singularity and Docker are supported). This is what we’ve been doing throughout these materials, when running the nf-core/viralrecon pipeline, where we used the option -profile singularity. With this option, Nextflow will download the necessary software containers to run each step of the pipeline, which are available in public repositories online.
To use Singularity and Docker containers, the respective programs have to be installed, which we detail in our software setup page.
As we’ve seen, the Mamba package manager and the containerisation solutions such as Docker and Singularity are trying to achieve similar things: enabling the reproducible installation of software and its dependencies in an isolated environment. So, why use one or the other?
Mamba is more user-friendly, allowing you to easily install packages of your choice. However, Mamba often works less well for more complex environments and can become extremely inneficient for environments with too many packages and conflicting versions. The recommendation is to keep your Mamba environments relatively small and atomic (e.g. an environment for each software package or for small sets of related packages).
Singularity and Docker, on the other hand, allow for more complex environments. However, to create your own container requires more advanced knowledge and a steep learning curve. Fortunately, there are many existing containers available for bioinformatics, which Nextflow uses in its pipelines.
15.4 Summary
- Linux is used in bioinformatics due to its open-source nature, powerful command-line interface, and compatibility with bioinformatics tools.
- Package managers such as
aptenable global software installations on Linux systems, simplifying the process of obtaining and managing software at the system level. - Local package managers such as
mambaallow users to install and manage software within user-specific environments, avoiding conflicts with system-level packages. - Software containers, such as Docker and Singularity, encapsulate entire environments in a file and can be used to manage more complex software enviroments.
- Bioinformatics workflow managers such as Nextflow can make use of software containers to run complex bioinformatic pipelines.