9 South Africa (Illumina)
This section demonstrates a start-to-finish analysis of a dataset sequenced on an Illumina platform, using the concepts and tools covered in previous sections. You can download the data from these links (two versions available):
- South Africa Case Study - Full Data – this includes data for 24 samples, which gives a more realistic sample size, but can take a few hours to run on a small computer.
- South Africa Case Study - Small Version – this includes data for a subset of 8 samples, which is more suitable for training purposes (but the results will look slightly different from the ones shown here).
By the end of this section, you should be able to:
- Prepare all the files necessary to run the consensus pipeline.
- Run the viralrecon pipeline to generate FASTA consensus from raw FASTQ files.
- Assess and collect several quality metrics for the consensus sequences.
- Clean output files, in preparation for other downstream analysis.
- Assign sequences to lineages using Nextclade and/or Pangolin.
- Contextualise your sequences in other background data and cluster them based on phylogenetic analysis.
- Integrate the metadata and results to generate useful visualisations of your data.
- Report your analysis.
We will analyse data from 24 samples collected in South Africa between Nov-Dec 2021. The samples were sequenced on an Illumina MiSeq platform.
The final product of our work (and main objective) is to produce a report of the analysis, which you can see here: South Africa Case Study Report.
In summary, the report addresses the following:
- What was the quality of the consensus sequences?
- What lineage/clade was each of our samples assigned to?
- How many clusters of samples did we identify?
- How did the detected lineages change over the time of sampling?
We also produce several essential output files, which would usually be necessary to upload our data to public repositories:
- Metadata (CSV)
- Consensus sequences (FASTA)
- Consensus sequence quality metrics (CSV)
- Variants (CSV)
9.1 Pipeline Overview
Our analysis starts with FASTQ files, which will be used with the nf-core/viralrecon Nextflow pipeline. This will give us several quality control metrics essential for our downstream analysis and reporting.
Critical files output by the pipeline will need to be further processed, including combining our consensus FASTA files and obtaining a list of filtered SNP/indel variants. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of mutations or variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions.
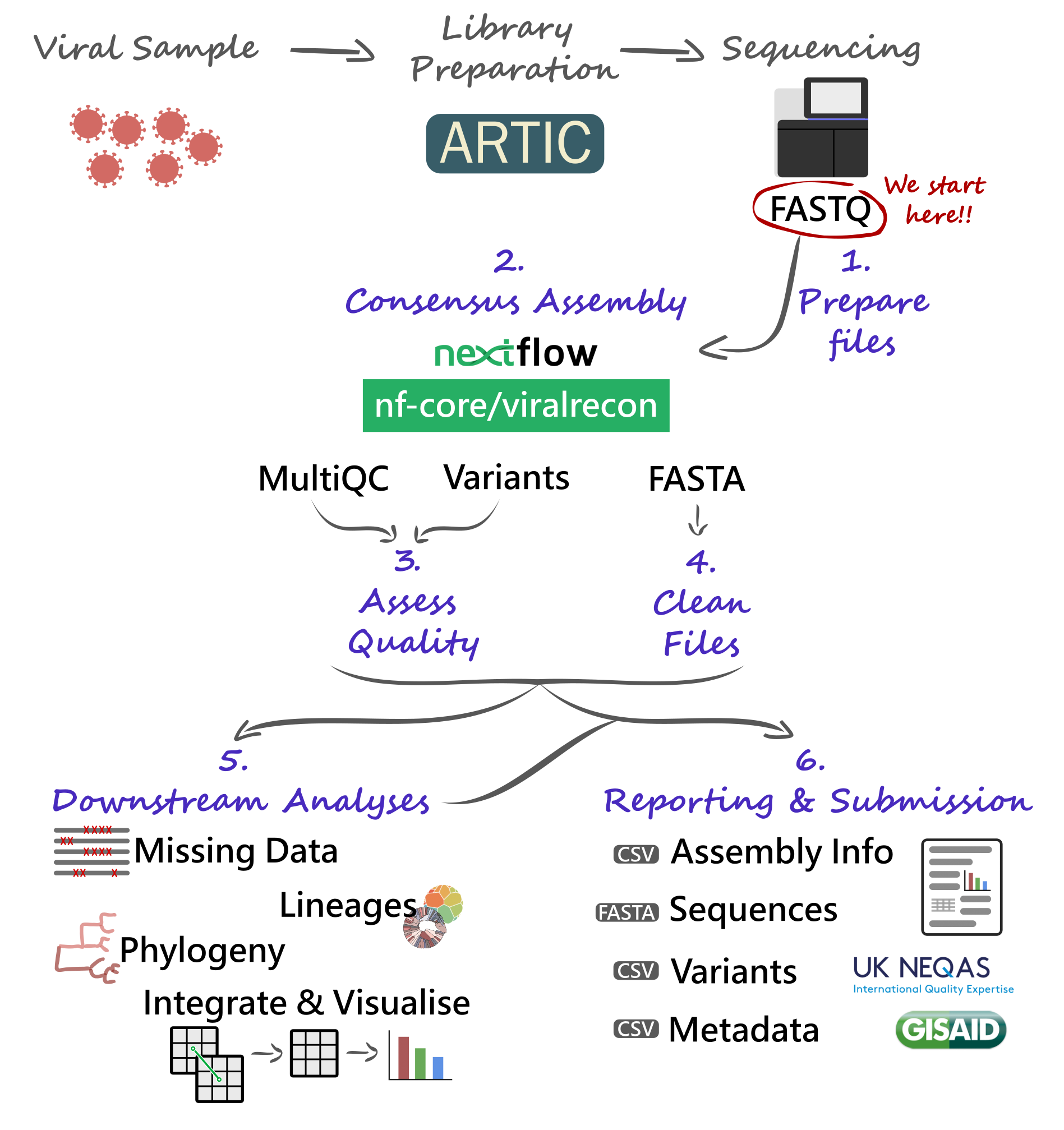
9.2 Preparing Files
Before we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. From the data we are starting with, we already have the following directories:
data→ contains the sequencing data in a sub-directory calledreads.resources→ files that were downloaded from public repositories.scripts→ bash and R scripts used to run the analysis.
We create two additional directories:
report→ files and documents that we report to our colleagues or upload to public repositories.results→ results of the analysis.
You can create directories from the command line using the mkdir command:
mkdir results
mkdir report9.2.1 Data
We start our analysis from FASTQ files generated by the Illumina sequencer. As this is paired-end sequencing, we have two files per sample (with suffix _1 and _2):
ls data/readsSRR17051908_1.fastq.gz SRR17051953_1.fastq.gz SRR17461700_1.fastq.gz SRR17712594_1.fastq.gz
SRR17051908_2.fastq.gz SRR17051953_2.fastq.gz SRR17461700_2.fastq.gz SRR17712594_2.fastq.gz
SRR17051916_1.fastq.gz SRR17054503_1.fastq.gz SRR17461712_1.fastq.gz SRR17712607_1.fastq.gz
SRR17051916_2.fastq.gz SRR17054503_2.fastq.gz SRR17461712_2.fastq.gz SRR17712607_2.fastq.gz
SRR17051923_1.fastq.gz SRR17088917_1.fastq.gz SRR17701832_1.fastq.gz SRR17712711_1.fastq.gz
SRR17051923_2.fastq.gz SRR17088917_2.fastq.gz SRR17701832_2.fastq.gz SRR17712711_2.fastq.gz
SRR17051932_1.fastq.gz SRR17088924_1.fastq.gz SRR17701841_1.fastq.gz SRR17712779_1.fastq.gz
SRR17051932_2.fastq.gz SRR17088924_2.fastq.gz SRR17701841_2.fastq.gz SRR17712779_2.fastq.gz
SRR17051935_1.fastq.gz SRR17088928_1.fastq.gz SRR17701890_1.fastq.gz SRR17712994_1.fastq.gz
SRR17051935_2.fastq.gz SRR17088928_2.fastq.gz SRR17701890_2.fastq.gz SRR17712994_2.fastq.gz
SRR17051951_1.fastq.gz SRR17088930_1.fastq.gz SRR17712442_1.fastq.gz SRR17712997_1.fastq.gz
SRR17051951_2.fastq.gz SRR17088930_2.fastq.gz SRR17712442_2.fastq.gz SRR17712997_2.fastq.gz9.2.2 Metadata
Metadata for these samples is available in the file sample_info.csv. Here is some of the information we have available for these samples:
sample→ the sample ID.collection_date→ the date of collection for the sample in the format YYYY-MM-DD.country→ the country of origin for this sample.geo_loc_region→ the region within the country where the sample was collected.latitude/longitude→ coordinates for sample location (in this case we’re only given a single coordinate for the whole country - in a real setting you may want to collect a precise location).sequencing_instrument→ the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).sequencing_protocol_name→ the type of protocol used to prepare the samples (e.g. ARTIC).amplicon_primer_scheme→ for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)
9.3 Consensus Assembly
See Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.
The first step in the bioinformatic analysis is to run the nf-core/viralrecon pipeline. But first we need to prepare our input files.
9.3.1 Samplesheet
For Illumina data, we need a samplesheet CSV file with three columns, indicating sample name (first column) and the respective FASTQ file paths for read 1 (second column) and read 2 (third column).
Because our FASTQ file names are not very user-friendly, we used some command-line tricks to help us produce this table:
# list read 1 files and save output in a temporary file
ls data/reads/*_1.fastq.gz > read1_filenames.txt
# list read 2 files and save output in a temporary file
ls data/reads/*_2.fastq.gz > read2_filenames.txt
# initiate a file with column names
echo "fastq_1,fastq_2" > samplesheet.csv
# paste the two temporary files together, using comma as a delimiter
paste -d "," read1_filenames.txt read2_filenames.txt >> samplesheet.csv
# remove the two temporary files
rm read1_filenames.txt read2_filenames.txtThese commands resulted in creating a file called samplesheet.csv, which contains the following:
head samplesheet.csvfastq_1,fastq_2
data/reads/SRR17051908_1.fastq.gz,data/reads/SRR17051908_2.fastq.gz
data/reads/SRR17051916_1.fastq.gz,data/reads/SRR17051916_2.fastq.gz
data/reads/SRR17051923_1.fastq.gz,data/reads/SRR17051923_2.fastq.gz
data/reads/SRR17051932_1.fastq.gz,data/reads/SRR17051932_2.fastq.gz
data/reads/SRR17051935_1.fastq.gz,data/reads/SRR17051935_2.fastq.gz
data/reads/SRR17051951_1.fastq.gz,data/reads/SRR17051951_2.fastq.gz
data/reads/SRR17051953_1.fastq.gz,data/reads/SRR17051953_2.fastq.gz
data/reads/SRR17054503_1.fastq.gz,data/reads/SRR17054503_2.fastq.gz
data/reads/SRR17088917_1.fastq.gz,data/reads/SRR17088917_2.fastq.gzSo, we programmatically created the last two columns of our file. We then opened this CSV file in Excel to add another column “sample” where we included our sample names and saved the file again as a CSV format. Here are the top few rows of the final file:
head samplesheet.csvsample,fastq_1,fastq_2
ZA01,data/reads/SRR17051908_1.fastq.gz,data/reads/SRR17051908_2.fastq.gz
ZA02,data/reads/SRR17051923_1.fastq.gz,data/reads/SRR17051923_2.fastq.gz
ZA03,data/reads/SRR17051916_1.fastq.gz,data/reads/SRR17051916_2.fastq.gz
ZA04,data/reads/SRR17051953_1.fastq.gz,data/reads/SRR17051953_2.fastq.gz
ZA05,data/reads/SRR17051951_1.fastq.gz,data/reads/SRR17051951_2.fastq.gz
ZA06,data/reads/SRR17051935_1.fastq.gz,data/reads/SRR17051935_2.fastq.gz
ZA07,data/reads/SRR17051932_1.fastq.gz,data/reads/SRR17051932_2.fastq.gz
ZA08,data/reads/SRR17054503_1.fastq.gz,data/reads/SRR17054503_2.fastq.gz
ZA09,data/reads/SRR17088930_1.fastq.gz,data/reads/SRR17088930_2.fastq.gz9.4 Consensus Quality
See Section 5.2, if you need to revise how to assess the quality of consensus sequences.
9.4.1 General Metrics
We used the MultiqQC report to assess the initial quality of our samples. The quality report can be found in results/viralrecon/multiqc/multiqc_report.html.
We paid particular attention to:
- Number of reads mapped to the reference genome.
- Median depth of coverage.
- Percentage of the genome with missing bases (‘N’).
- Number of SNP + Indel variants.
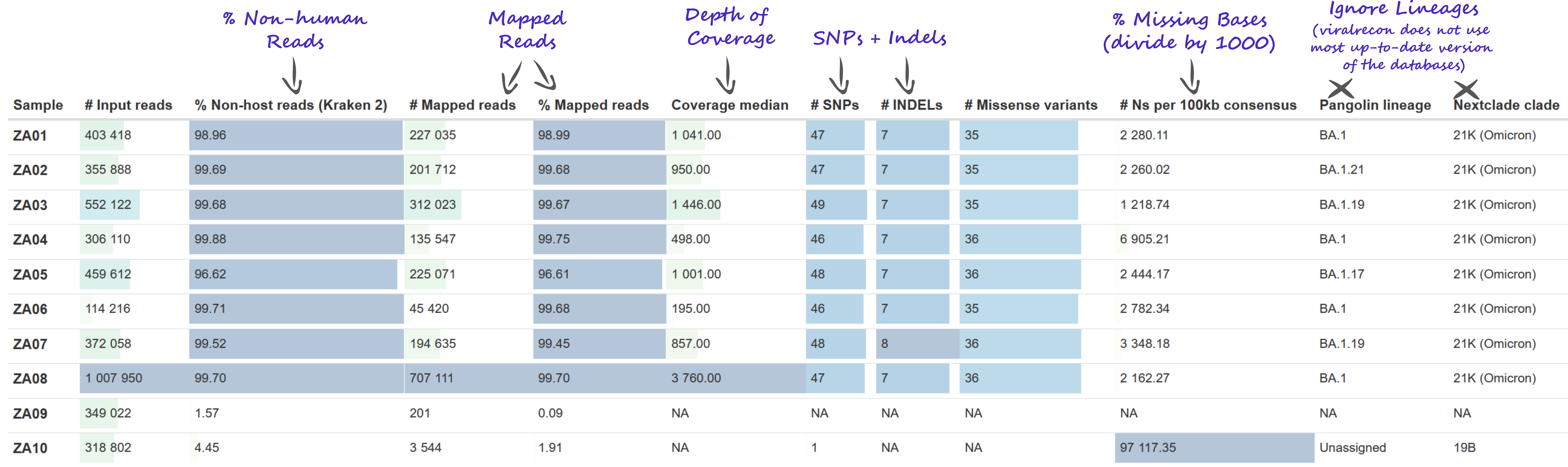
We noted that:
- 2 samples - ZA09 and ZA12 - completely failed. This was already noted after we ran the pipeline, above. The reason seems to be because these samples have a very low percentage of non-human reads (less than 2%), indicating that the sequenced material was mostly human. Because of this, there were not enough reads to assemble a genome.
- 2 other samples - ZA10 and ZA23 - had > 90% missing bases, also indicating a very poor assembly. The reason was the same as above, both samples had low % of non-human reads.
- 1 samples - ZA14 - had ~39% missing bases. In this case the reason was not a low number of mapped reads. For example, sample ZA13 had a very similar median depth of coverage (121 vs 161 in ZA14) and even fewer mapped reads (24k vs 60k in ZA14). But ZA13 only had 2% missing bases. However, upon inspection of the amplicon heatmap, we detected that several amplicons were not properly amplified in ZA14 compared to ZA13. Therefore, the reason for high % missing bases in ZA14 was due to low amplification efficiency in this sample.
- There was some systematic dropout for some amplicons, in particular
nCoV-2019_64had very low amplification in several of the samples. Of note was alsonCoV-2019_73, and other neighbouring amplicons.

Besides the MultiQC report, the pipeline also outputs a CSV file with collected summary metrics (equivalent to the first table on the report): results/viralrecon/multiqc/summary_variants_metrics_mqc.csv. We will use this file later to join this information with our metadata and lineage assignment using the R software (detailed in “Integration & Visualisation” section, below).
9.4.2 Variants
We also looked at the table of variants obtained from the pipeline. This is output in results/viralrecon/variants/ivar/variants_long_table.csv. This table can be very useful to keep track of particular mutations that may be increasing over time. Later, we will tidy this table to attach to our reported results (“Integration & Visualisation” section).
But for now, we will explore this table to address a few more quality-related questions. We opened this table in Excel to answer the following:
- Where there samples with a high number of intermediate allele frequencies? This could indicate mixed samples due to cross-contamination.
- Where there samples with frameshift mutations? These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.
By manual inspection of this table (and using the “filter” feature in Excel), we found 74 variants with alternative allele frequency less than 75%. These were spread across samples, all samples having less than 10 such low-frequency mutations, and most samples having less than 5. Sample ZA23 - which we had previously highlighted has having a high % missing bases (39%) - had 8 low-frequency mutations out of a total of 48 mutations in this sample (~16%), suggesting further quality issues in this sample.

9.5 Clean FASTA
The pipeline outputs the consensus sequences in results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa (one file for each sample). For downstream analysis, it is convenient to combine all these sequences into a single file, and also clean the sequence names (to remove some text – ” MN908947.3” –, which is added by the bcftools variant caller).
We created a new script to clean our consensus FASTA files, which we ran with bash scripts/02-clean_fasta.sh:
# combine and clean FASTA files
cat results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa | sed 's| MN908947.3||' > report/consensus.faThis command does two things:
- Combine all our FASTA consensus sequences into a single file (using
cat). - Clean the sequence names (using
sed).
The output was saved as a new FASTA file: report/consensus.fa.
9.5.1 Missing Intervals
As a further quality check, we also generated a table of missing intervals (indicated by the N character in the FASTA sequences). We used the seqkit software to achieve this.
First, we activate our software environment:
mamba activate seqkitThen, we ran the script bash scripts/03-missing_intervals.sh, which includes the following command:
seqkit locate -i -P -G -M -r -p "N+" report/consensus.fa > results/missing_intervals.tsvThis software outputs a tab-delimited table, which we saved as results/missing_intervals.tsv. The table looks like this (only the top few rows are shown):
seqID patternName pattern strand start end
ZA01 N+ N+ + 1 54
ZA01 N+ N+ + 22771 22926
ZA01 N+ N+ + 23603 23835
ZA01 N+ N+ + 26948 26948
ZA01 N+ N+ + 26968 27137
ZA01 N+ N+ + 29801 29867
ZA02 N+ N+ + 1 54
ZA02 N+ N+ + 22771 22921
ZA02 N+ N+ + 23603 23835We opened this file missing_intervals.tsv in Excel and quickly calculated the length of each interval. We noted that two samples - ZA10 and ZA23 - both have a continuous interval of over 18kb missing bases, which is not surprising as we had already identified these samples has having >90% missing data. We make a note of these samples as being possibly problematic in downstream analysis steps.
9.6 Downstream Analyses
Based on the clean consensus sequences, we then perform several downstream analysis.
9.6.1 Lineage Assignment
See Section 6.1, if you need to revise how lineage assignment works.
Although the Viralrecon pipeline runs Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). An up-to-date run of both of these tools can be done using each of their web applications:
However, for automation, reproducibility and traceability purposes, we used the command line versions of these tools, and included their analysis in two scripts.
For Nextclade, we first activate the software environment:
mamba activate nextcladeAnd then we ran the script bash scripts/04-nextclade.sh, which contains the following commands:
# get nextclade data
nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
# run nextclade
nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.faThe first command downloads the latest version of the Nextclade background data using nextclade dataset. We use that data as input to the second command (nextclade run) to make sure it runs with the most up-to-date lineages.
For Pangolin, we first activate the software environment:
mamba activate pangolinAnd then we ran the script bash/04-pangolin.sh, which contains the following commands:
# update pangolin data
pangolin --update-data
# run pangolin
pangolin --outdir results/pangolin/ --outfile pango_report.csv report/consensus.faSimilarly to before, we first ran pangolin --update-data to ensure we were using the latest lineages available. We can check the version of the data used with pangolin --all-versions (at the time we ran this we had pangolin-data: 1.23.1).
Both of these tools output CSV files, which can be open in Excel for further examination.
Opening the pangolin results (results/pangolin/pango_report.csv), we noticed that the two problematic samples – ZA10 and ZA23 – failed the QC due to high fraction of missing data. The other samples all seemed to have been assigned to “Omicron” variant with a high support.
Opening the nextclade results (results/nextclade/nextclade.tsv), we noticed that the two problematic samples were classified as “recombinant”! We know from our quality control that we should not trust this assessment, and that most likely these are bad quality samples, not true recombinant lineages. Nextclade is more relaxed in assigning samples to lineages, so we should always check the QC status as well. We will notice that both of these samples were assigned QC status “bad”, due to their high percentage of missing data (nextclade uses a stringent threshold of 3000 sites, or ~10%, missing data). Two other samples had “bad” QC status. Sample ZA14 due to high % of missing data, and sample ZA18 from a mixture of a high number of private mutations and the presence of a frameshift mutation in ORF1b.
Like before, we will do further analysis (and visualisation) of these data using the software R, in the section “Integration & Visualisation”, detailed below.
9.6.2 Phylogeny
See Section 7.1, if you need to revise how to build phylogenetic trees.
Although a tool such as Nextclade can place our samples in a global phylogeny context, sometimes it may be convient to build our own phylogenies. This requires three steps:
- Producing a multiple sequence alignment from all consensus sequences.
- Tree inference.
- Tree visualisation and annotation.
Before our analysis, we first activated our software environment:
mamba activate phyloWe performed the first two steps with the following script, which we ran with bash scripts/05-phylogeny.sh:
# alignment
mkdir -p results/mafft
mafft --6merpair --maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa
# tree inference
mkdir -p results/iqtree
iqtree2 -s results/mafft/alignment.fa --prefix results/iqtree/consensusThe output of iqtree includes a tree file, which can be visualised using FigTree (or online using Microreact). The figure below shows our tree, which shows all our samples fall mostly clustering together. This makes sense, as all our samples were classified as “Omicron (BA.1-related)”.
It is worth noting that the samples ZA10, ZA14 and ZA23 are not included in this phylogeny, as they contained >20% missing data (we used that threshold with MAFFT alignment, option --maxambiguous 0.2). Also, note that sample ZA18, which Nextclade had identified as “bad” QC (due to excess private mutations) also appears slightly separated from the other samples in the tree (the sample in red on the tree). The sample still clusters well with the rest, suggesting we can probably trust that it is indeed an Omicron variant, however the excess of private mutations (which may be due to sequencing errors) is making it stand apart from the others in the phylogeny.
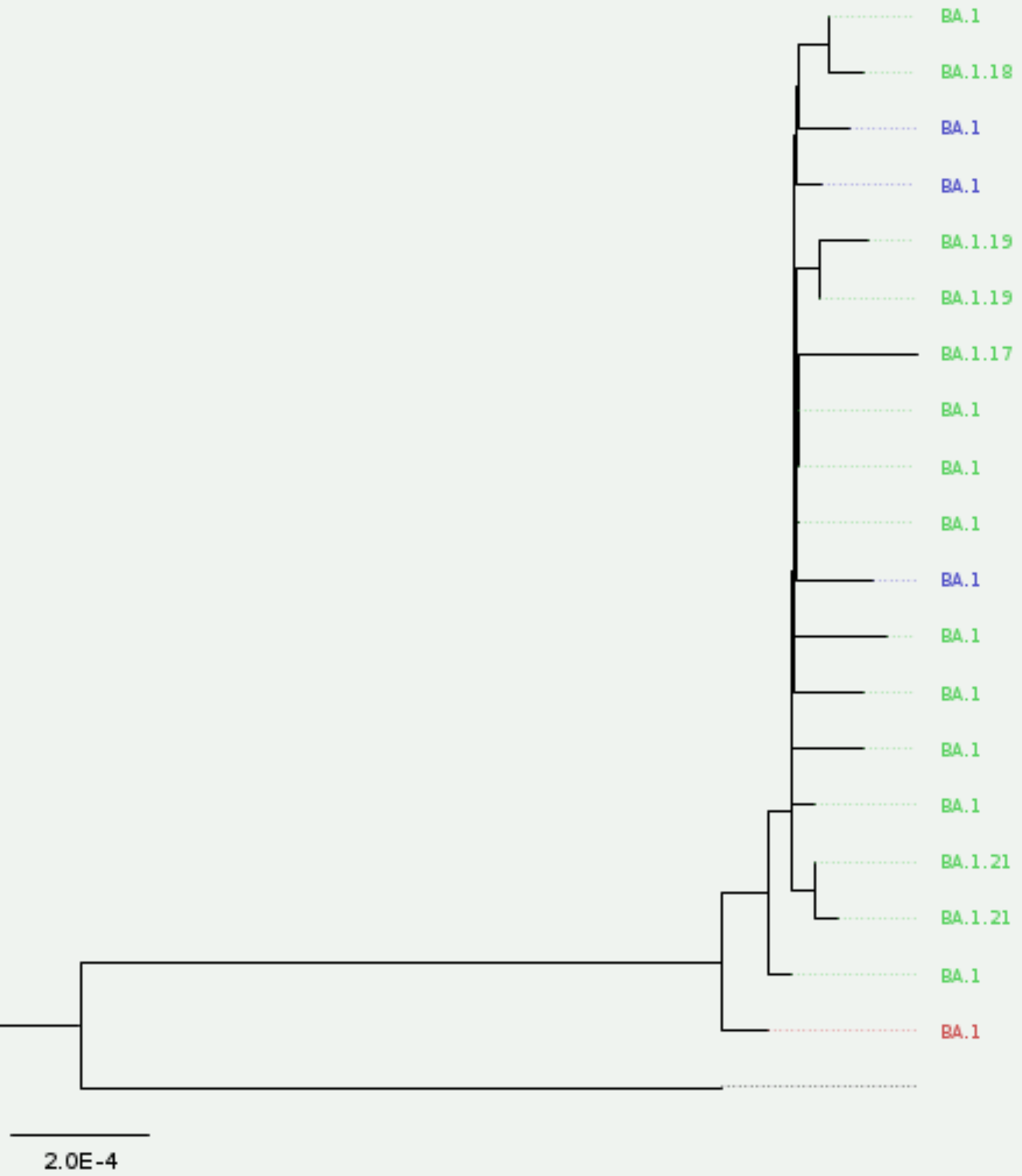
9.6.3 Clustering
We identified groups of similar sequences in our data using the software civet (Cluster Investigation and Virus Epidemiology Tool). This software compares our samples with a background dataset of our choice, which givus us more context for our analysis. In this case we are using the example background data that comes with civet. However, in a real-world analysis, it would have been ideal to choose local samples as background data. For example, we could download samples from South Africa from around the time period of our sample collection, from GISAID following the instructions on the civet documentation (you need an account on GISAID to obtain these data).
For this example, we already prepared civet background dataset saved in resources/civet_background_data.
Before our analysis, we first activate our software environment:
mamba activate civetThen, we ran the script bash scripts/06-civet.sh, which contains the following code:
# run civet analysis
civet \
-i sample_info.csv \
-f report/consensus.fa \
-icol sample \
-idate collection_date \
-d resources/civet_background_data/ \
-o results/civetThe result of this analysis includes an interactive HTML report (in results/civet/civet.html). We can see that our samples were grouped into a single catchment. This makes sense from our previous lineage/variant analysis: all our samples were classfied as Omicron VOC. We can see that our samples seem to be more diverged from the Australian BA.1 sample present in the background data, with new SNPs in our samples creating a longer branch in the tree.

Civet also outputs a CSV file (results/civet/master_metadata.csv), which includes the catchment that each sample was assigned to. We will use this CSV file later to integrate this information with other parts of our analysis, in R, detailed in the “Integration & Visualisation” section.
9.7 Integration & Visualisation
At this point in our analysis, we have several tables with different pieces of information:
sample_info.csv→ the original table with metadata for our samples.results/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv→ quality metrics from the MultiQC report generated by the viralrecon pipeline.results/nextclade/nextclade.tsv→ the results from Nextclade.results/pangolin/pango_report.csv→ the results from Pangolin.results/civet/master_metadata.csv→ the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.
To consolidate our analysis, we tidied and integrated the information from across these different files, into a single table using the software R. The script used to do this is in scripts/07-data_integration.R. Because this is an R script, we opened it in RStudio to execute the code.
The output of our script is a new tab-delimited table, which we saved in report/consensus_metrics.tsv, and contains the following columns:
sample→ sample ID.collection_date→ date of collection day.collection_week→ date of collection week (useful for summarising/visualising counts per-week).country→ country of origin.latitude/longitude→ latitude and longitude of collection.n_mapped_reads→ number of mapped reads.median_depth→ median depth.pct_missing→ percentage of missing data.pct_coverage→ percentage of coverage.n_variants→ number of SNP + indel variants detected.nextclade→ nextclade clade.qc_status→ QC status as determined by Nextclade (“bad”, “mediocre”, “good”).lineage→ Pangolin lineage.who_variant→ variant of concern designation.catchment→ catchment group from Civet.
This table, which aggregates information from many of the tools we used, was then used to produce different visualisations of our analysis. These visualisations were also done using the R software (scripts/08-visualisation.R), and integrated into a report, shown below.
9.8 Bonus: Full Workflow
Although we have ran each of the steps of our analysis individually (each in their own script), now that we have everything working, we could integrate all these steps into a single “master” script:
#!/bin/bash
# make mamba activate command available
eval "$(conda shell.bash hook)"
source $(mamba info --base)/etc/profile.d/mamba.sh
# make report directory
mkdir -p report
mamba activate nextflow
# run viralrecon
nextflow run nf-core/viralrecon \
-r 2.6.0 -profile singularity \
--max_memory '15.GB' --max_cpus 8 \
--platform illumina \
--input samplesheet.csv \
--outdir results/viralrecon \
--protocol amplicon \
--genome 'MN908947.3' \
--primer_set artic \
--primer_set_version 3 \
--skip_assembly --skip_asciigenome \
--skip_pangolin --skip_nextclade
# combine and clean FASTA files
cat results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa | sed 's| MN908947.3||' > report/consensus.fa
mamba activate seqkit
# create missing bases TSV file
seqkit locate -i -P -G -M -r -p "N+" report/consensus.fa > results/missing_intervals.tsv
mamba activate nextclade
# get nextclade data
nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
# run nextclade
nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa
mamba activate pangolin
# update pangolin data
pangolin --update-data
# run pangolin
pangolin --outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa
mamba activate phylo
# alignment
mkdir -p results/mafft
mafft --6merpair --maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa
# tree inference
mkdir -p results/iqtree
iqtree2 -s results/mafft/alignment.fa --prefix results/iqtree/consensus
# data integration and cleaning
Rscript scripts/07-data_integration.RNotice that we included the R script that does the data cleaning here, using the Rscript program that allows to execute an R script from the command-line.
Having this “master” script, we could run all these steps from start-to-finish with a single command, which can be very useful if you want to fully automate your analysis across multiple runs.