3 Visualising structures with ChimeraX
- Familiarise yourself with the ChimeraX interface.
- Load and import solved protein structures.
- Perform basic manipulations: rotate, zoom, colour, and highlight.
- Select residues, chains, and atoms using the selection syntax.
- Import data from UniProt and combine it with structural information.
- Save and manage ChimeraX sessions for later use.
3.1 Overview
ChimeraX provides a graphical user interface (GUI) and a command line interface (CLI). You can combine both for flexible and efficient structure visualisation.
3.2 Opening a structure
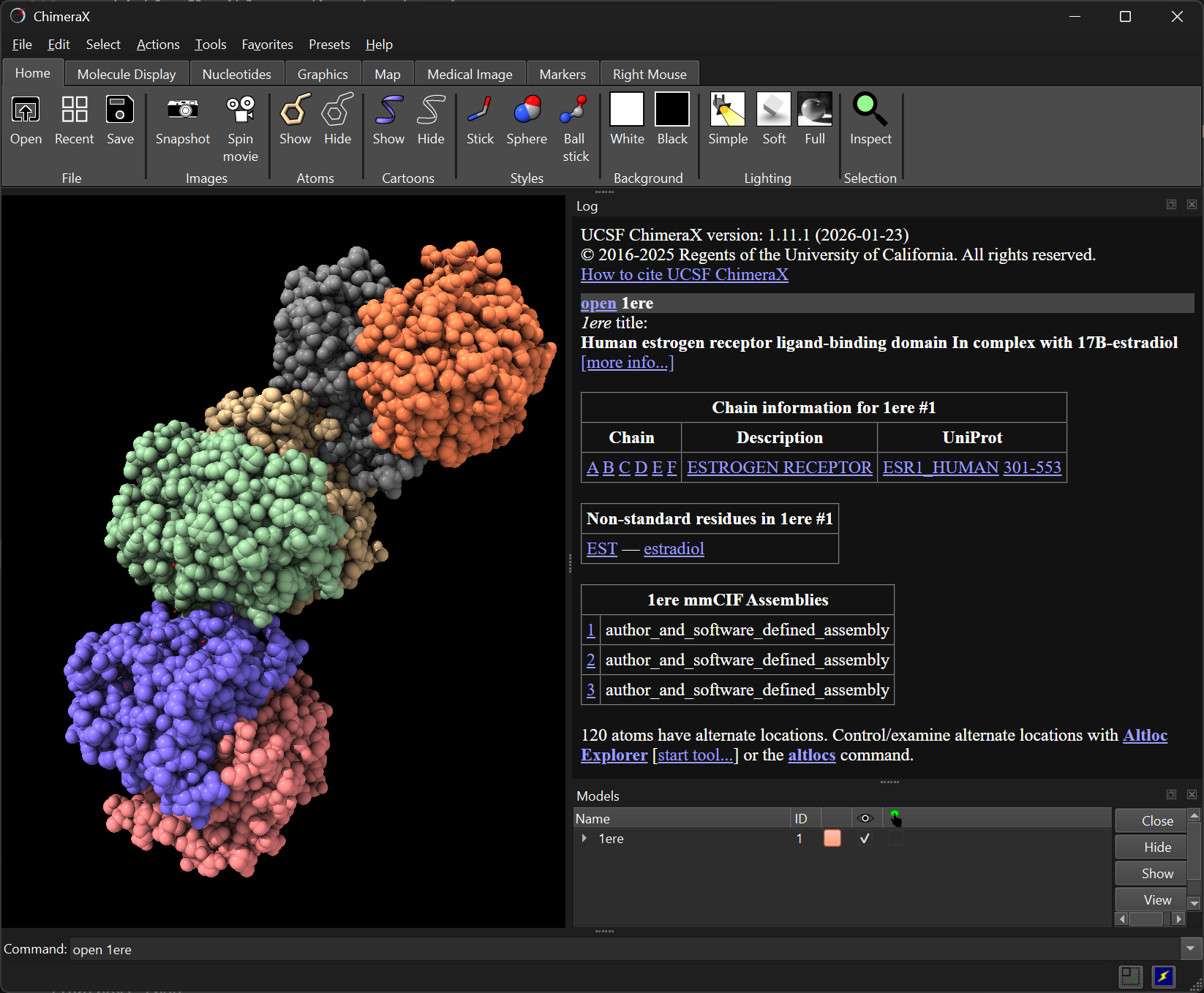
To open a PDB file from the command line, use open. For example, to load the ligand-binding domain of the human Estrogen Receptor (PDB: 1ERE):
open 1ereThe structure appears in the main window. Use the mouse to rotate, zoom, and pan.
3.3 Inspecting the structure
Use the info command to obtain information about the loaded models:
info2 models
#1, 1ere, shown
11376 atoms, 11472 bonds, 1530 residues, 6 chains (A,B,C,D,E,F)
12 missing structure
#1.1, missing structure, shown, 12 pseudobonds- Model #1: main structure (11376 atoms, 11472 bonds, 1530 residues, 6 chains).
- Model #1.1: missing residues (represented as pseudobonds).
To get details about chains:
info polymersChimeraX lists residue ranges for each chain. For 1ERE, each chain has three segments (305–330, 337–461, 465–548), repeated because this PDB entry consists of three assemblies of a homodimer (so, 3 x 2 = 6 chains).
To view metadata such as ligands:
log metadata| Metadata for 1ere#1 | |
|---|---|
| Title | Human estrogen receptor ligand-binding domain In complex with 17Β-estradiol |
| Citation | Brzozowski, A.M., Pike, A.C., Dauter, Z., Hubbard, R.E., Bonn, T., Engstrom, O., Ohman, L., Greene, G.L., Gustafsson, J.A., Carlquist, M. (1997). Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389, 753-758. PMID: 9338790. DOI: 10.1038/39645 |
| Non-standard residue | EST — estradiol |
| Gene source | Homo sapiens (human) |
| Experimental method | X-ray diffraction |
| Resolution | 3.1Å |
For example, this shows the presence of residues named EST, corresponding to the Estradiol ligand included with this structure.
To list chains and their UniProt references:
log chains| Chain | Description | UniProt |
|---|---|---|
| A–F | Estrogen Receptor | ESR1_HUMAN 301–553 |
3.4 Selecting items
Selections are central in ChimeraX, and can be used to highlight, manipulate, or modify specific parts. Selections are highlighted in the viewer and can be modified or combined.
Selections use a target specifier syntax (docs), including:
#→ model number/→ chain ID (uppercase letters):→ residue numbers@→ atom names
Examples:
Select model #1:
select #1Select chain A:
select /ASelect residue 500 in chain A:
select /A:500Select alpha carbon of residue 305:
select /A:305@CASelect residues 400–500 in chain A:
select /A:400-500Clear selection:
select clearSelect all:
select all
3.4.1 Combining and modifying selections
Selections can be built incrementally with select add and select subtract. Examples:
Select all residues in chain A except for residues 400 to 500:
select /A select subtract /A:400-500Add back residues 400 to 410:
select add /A:400-410
Selections can be combined using logical operators:
&(AND)|(OR)
Examples:
Select residues 305–330 in either chain A or chain F:
select /A:305-330 | /F:305-330Select all serine (SER) residues in a range:
select /A:305-330 & :SER
In the last example, note how we used the aminoacid name (SER) instead of the residue number to select all serine residues in that range.
3.4.2 Select keywords
There are convenience keywords that can be used to select common groups of atoms:
| Keyword | What it selects |
|---|---|
protein |
all protein atoms |
nucleic |
all DNA/RNA atoms |
ligand |
non-polymer atoms (small molecules, ions) |
solvent |
water molecules |
sel |
currently selected elements |
Residues are also annotated with names. We’ve already seen how we can select all serine residues by their name using :SER.
Ligands are usually also named. For example, in our structure, the ligand (estradiol) is annotated with the keyword EST, so we can select it with:
select :ESTAlternatively, we could use the generic keyword:
select ligandThe latter would select every ligand in the loaded structures, so if there was more than one ligand, all of them would be selected this way.
Zooming and rotation
Zoom to a selection with view:
select /A:305@CA
view selSet the centre of rotation on a certain item with cofr. For example, set the centre of rotation on the ligand:
cofr :EST3.5 Delete objects
You can use the delete command to remove parts of your struture from view. In our example, the 1ERE structure contains 6 repeated chains (A-F). To only keep chain A and delete the rest:
select #1/A
delete atoms ~sel- First select chain A (
/A) from the main model (#1). - Then delete everything that is not selected:
~sel(the~symbol negates the selection).
The same could have been achieved with:
delete #1/B-F3.6 Naming and saving selections
Selections can be saved for reuse.
For example, we save a selection of residues that are commonly mutated in cancers (Fuqua, Gu & Rechoum, 2015):
select /A:536,537,538
name frozen muts /A:536,537,538Now you can refer to muts:
# zoom-in on this selection
view muts
# select these residues
select muts
# select everything except these residues
select ~muts
# colour these residues red
color muts red3.7 Sequence viewer
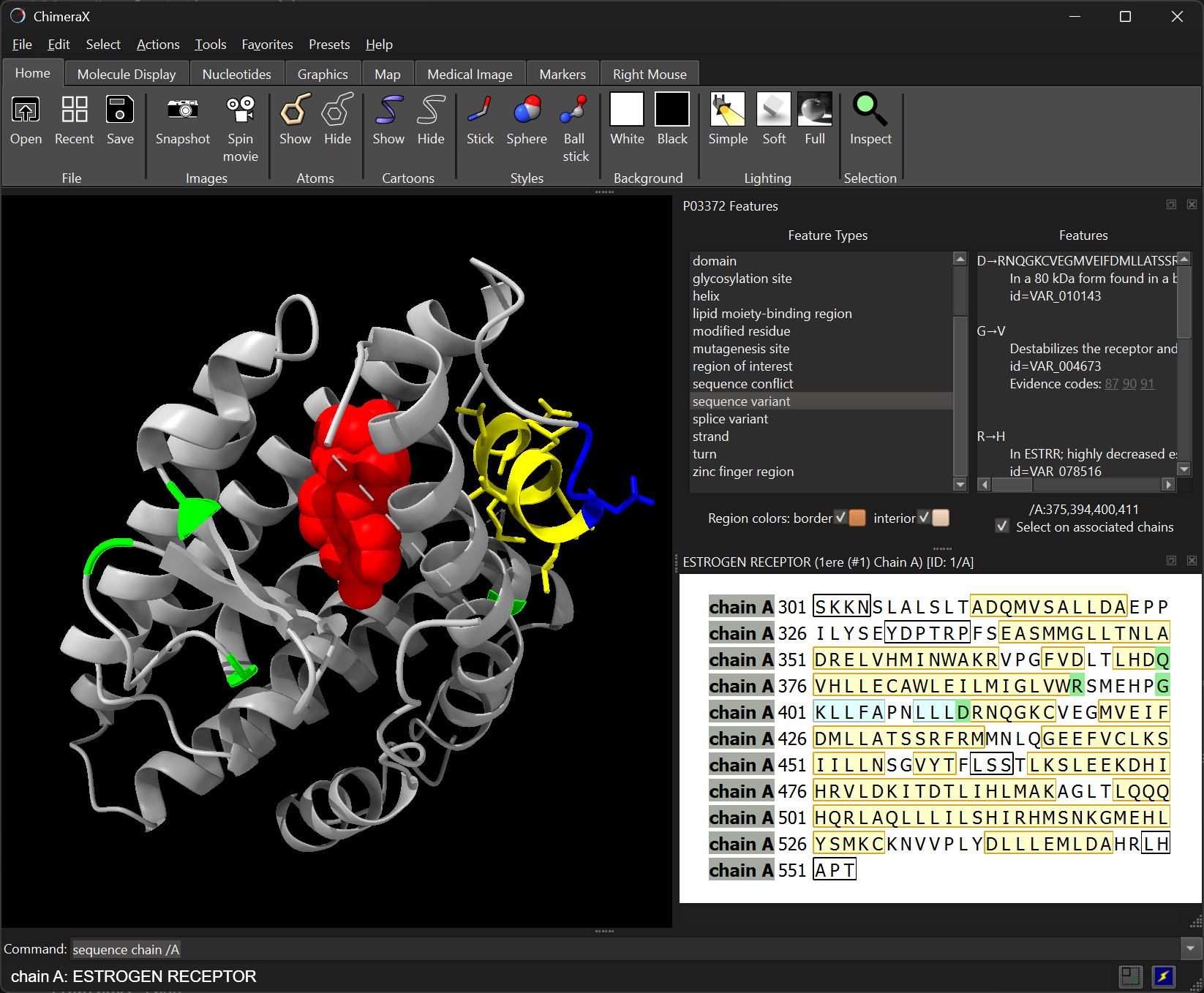
Open a sequence window to visualise and select residues:
sequence chain /A
Click residues in the sequence to select them in 3D.
Selected residues are highlighted in green.
Secondary structure is colour-coded: α-helix (yellow), β-strand (blue).
Missing residues appear in black boxes.
Residue numbers correspond to the UniProt numbering.
Selected residues are highlighted in green.
Secondary structure is colour-coded: β-strand in blue and α-helix in yellow.
Missing residues appear in black boxes. These often correspond to disordered regions or flexible termini that could not be resolved in the protein structure.
3.8 Structural representations
Proteins can be represented visually in different ways.
Cartoon representation highlights secondary structure:
hide atoms show cartoonAtomic representation shows all atoms:
hide cartoon show atomsAlternate atomic styles can be used:
hide cartoon show atoms style stick style sphere style ballMix representations can be used. For example, cartoon for protein, spheres for ligand:
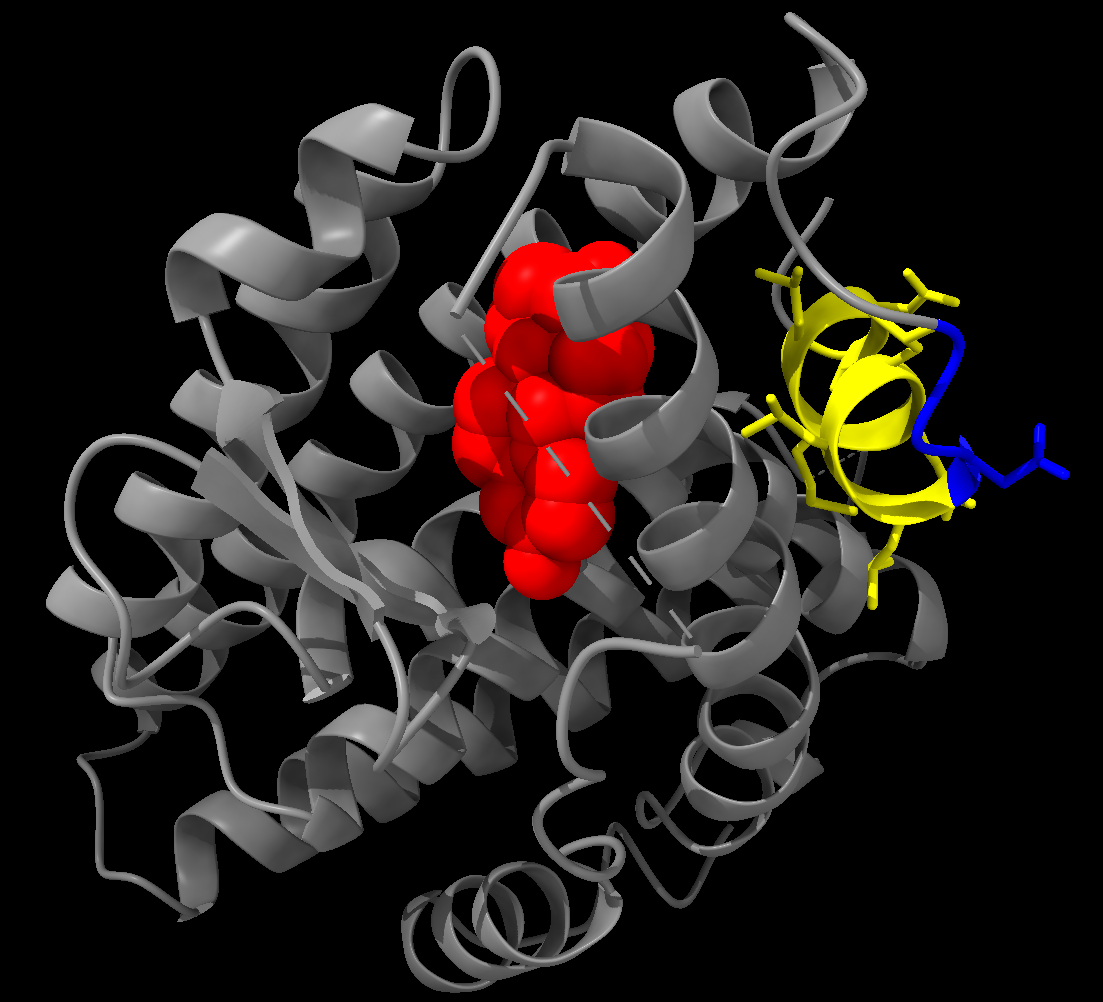
hide atoms show cartoon show :EST style :EST sphereColouring example:
color protein grey color :538-546 yellow show :538-546 atoms style :538-546 stick color muts blue color :EST red- Colour the protein grey.
- Colour residues 538-546 yellow, and show their atoms using a stick style.
- Colour the
mutsselection blue. - Colour the ligand (EST) red.
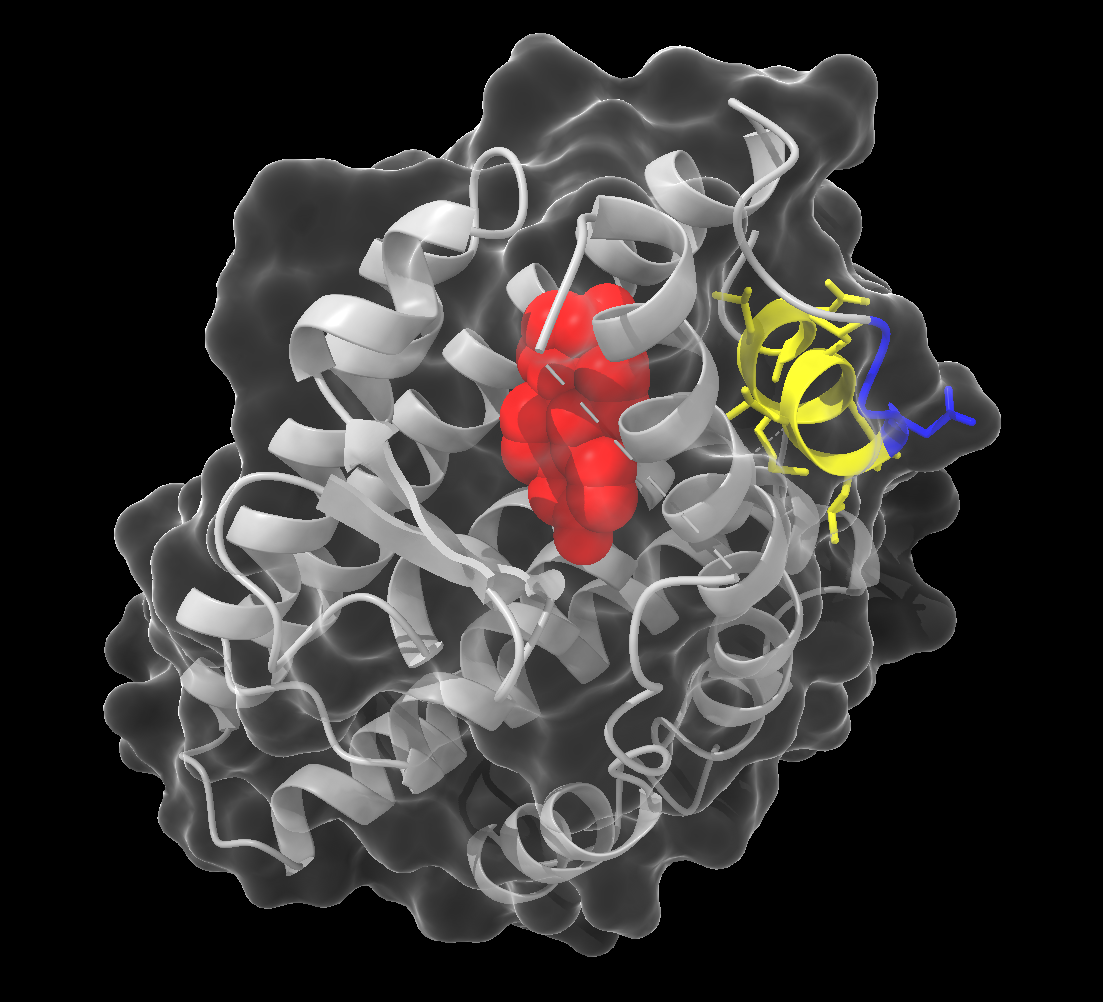
3.9 Surface representation
You can also represent structures using surfaces, which can help visualise the shape of the molecule.
Example:
surface protein color white transparency 80This command creates a surface representation of the protein, coloured white with 80% transparency. We keep the cartoon representation, allowing us to view the secondary structure inside the surface.
To hide the surface:
surface hide3.10 Adding UniProt annotations
Structural information is often easier to interpret alongside functional annotations. ChimeraX can retrieve UniProt annotations directly.
For ESR1, the corresponding UniProt entry is P03372. We can open its annotations with:
open P03372 from uniprot format uniprotThis opens a sequence annotation panel showing features such as:
- functional domains
- transmembrane regions
- experimentally observed variants
- binding sites
Clicking a feature in the panel will highlight the corresponding residues in the structure.
Combining structural and functional annotations helps identify regions likely to be sensitive to mutation.
3.11 Saving sessions and structures
Save a ChimeraX session by changing to the destination directory, and use the save command:
cd ~/Course_Materials/
save 1ERE_annotated.cxs.cxs is a ChimeraX-specific format. To reopen the session:
cd ~/Course_Materials/
open 1ERE_annotated.cxsThe save command can also be used to save protein structures in standard formats:
save 1ERE_annotated.pdb
save 1ERE_annotated.cifThis allows using ChimeraX to convert between struture formats: open a .pdb file and save as .cif, or vice-versa.
3.12 Exercises
Start a new session with chain A of 1ERE (Human estrogen receptor):
close
open 1ere
delete #1/B-FTry to recreate the visualisation shown below, where:
- The protein is shown in cartoon representation using
royalbluecolour. - The ligand (EST) is shown in sphere style using
greycolour for carbon atoms andmagentafor oxygen atoms. - The key protein residues for binding the ligand - Glu353, Arg394, His524, Leu525 - are highlighted in
goldcolour and their atoms are shown in ball style. - Zoom in specifically on these four residues using the
viewcommand.
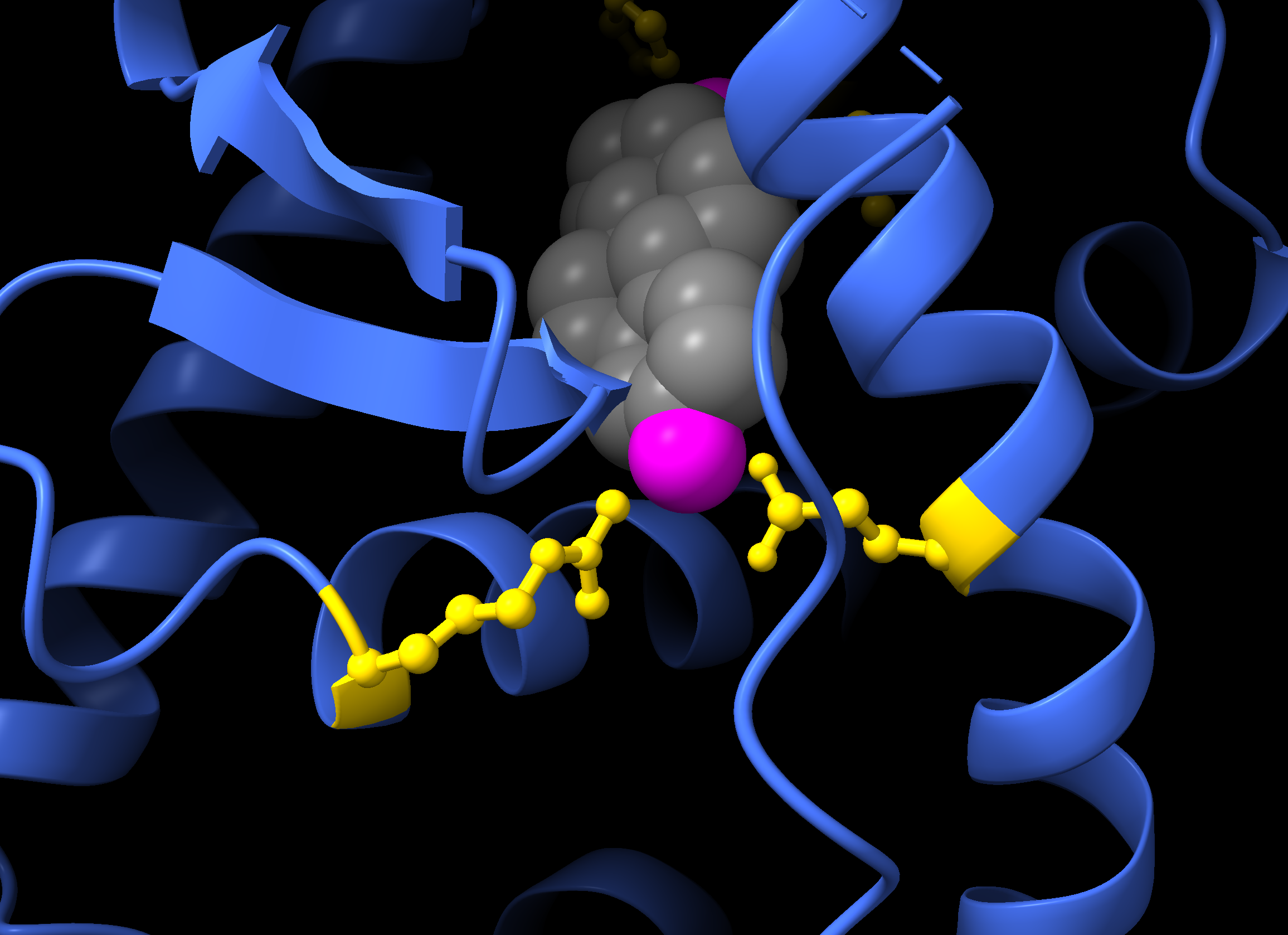
Try to recreate this view of the murine AA amyloid fibril (PDB: 6DSO):

Try and recreate this RCSB-PDB molecule of month view (PDB: 1AOI):

Make sure to colour each DNA chain slightly differently. You can use log chains to find out which chains correspond to the protein and which are the DNA.
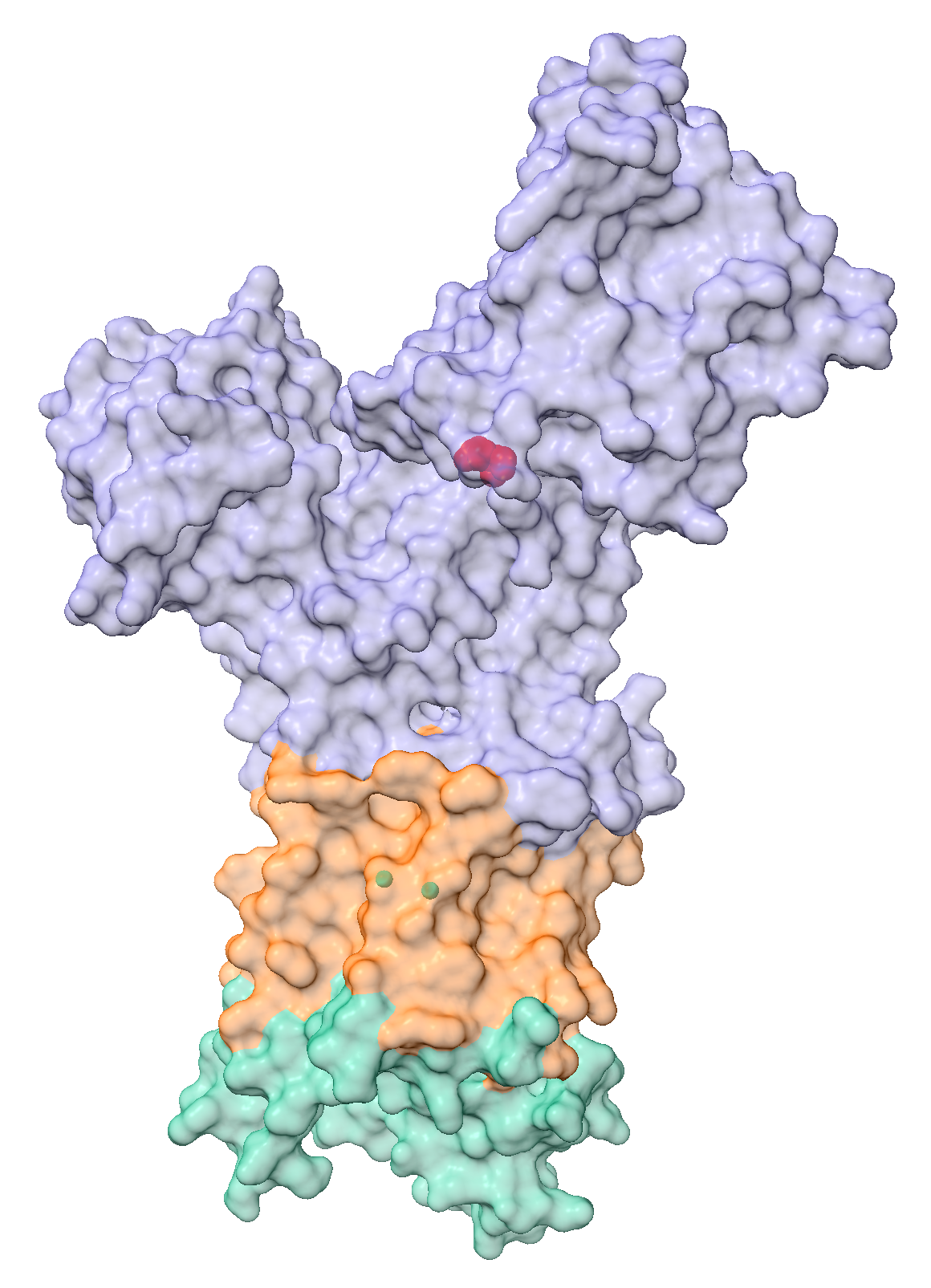
Try to create a similar view to the following RBSB-PDB molecule of the month (PDB: 1SU4).

It might be hard to recreate exactly the same view, but here are some guidelines for what you could try:
- Hide the atoms
- Draw a surface representation of the protein using a transparency of 60%
- Show the calcium ion and colour it cyan
- Use
log metadatato see how the Calcium molecule is called
- Use
- Import metadata from UniProt (you might have to go to PDB to find the UniProt ID). This should allow you to select:
- Active site: Show as atoms and colour red
- Topological domain lumenal: Colour
#1b9e77 - topological domain cytoplasmic: Colour
#7570b3 - transmembrane region: Colour
#d95f02
We start by opening the structure and hiding the atoms, then showing the surface representation of the protein with a transparency of 60%, and showing the CA atoms coloured cyan:
close
open 1SU4
hide cartoon
hide atoms
surface protein transparency 60
show :CA
colour :CA cyanWe import metadata from UniProt, using the UniProt ID P04191, which we can find in the PDB entry for 1SU4:
open P04191 from uniprot format uniprotWe select each of the features indicated, and colour them accordingly. Here, we give the exact residues, which we copied from the log window, as we clicked on each of the UniProt annotations.
select /A:70-89,274-295,778-787,852-897,950-964
colour sel #1b9e77
select /A:1-48,111-253,314-757,809-828,918-930,986-994
colour sel #7570b3
select /A:49-69,90-110,254-273,296-313,758-777,788-808,829-851,898-917,931-949,965-985
colour sel #d95f02
select /A:351
show sel atoms
style sel sphere
colour sel red
select clearThis should give you the following view:
3.13 Summary
At first, ChimeraX may look like a graphics program, however its real power comes from its range of commands and flexible selection syntax.
ChimeraX combines a graphical interface and command line interface.
Structures are loaded with the
opencommand. ChimeraX can fetch structures directly from the PDB using their identifier.Structural information can be inspected using
infocommands. These commands reveal models, chains, residues, ligands, and experimental metadata.ChimeraX uses a concise syntax to specify models (
#), chains (/), residues (:), and atoms (@).Selections can be modified and combined. Commands such as
add,subtract,&, and|allow complex selections.Structures can be displayed in different representations:
- Cartoon views emphasise secondary structure
- Atom-based views show detailed interactions
- Surface views reveal overall shape and interfaces
Saving selections allows you to refer to important residues or regions later.
ChimeraX sessions can be saved and reopened. This allows you to preserve visualisations and annotations for later work.
ChimeraX documentation
ChimeraX User Guide https://www.cgl.ucsf.edu/chimerax/docs/user/index.html
ChimeraX Tutorials https://www.cgl.ucsf.edu/chimerax/tutorials.html