library(ggResidpanel)
library(performance)11 Checking assumptions
Learning outcomes
- Know what statistical assumptions apply to logistic regression (and GLMs)
- Be able to evaluate whether a logistic regression meets the assumptions
11.1 Libraries and functions
Click to expand
import pandas as pd
from plotnine import *
import statsmodels.api as sm
import statsmodels.formula.api as smf
from scipy.stats import *
import matplotlib.pyplot as plt
import numpy as np
from statsmodels.stats.outliers_influence import variance_inflation_factor
from patsy import dmatrix11.2 Assumptions
Although generalised linear models do allow us to relax certain assumptions compared to standard linear models (linearity, equality of variance of residuals, and normality of residuals).
However, we cannot relax all of them - some key assumptions still exist.
11.2.1 Assumption 1: Distribution of response variable
Although we don’t expect our response variable \(y\) to be continuous and normally distributed (as we did in linear modelling), we do still expect its distribution to come from the “exponential family” of distributions.
The exponential family
The exponential family contains the following distributions, among others:
- normal
- exponential
- Poisson
- Bernoulli
- binomial (for fixed number of trials)
- chi-squared
You can use a histogram to visualise the distribution of your response variable, but it is typically most useful just to think about the nature of your response variable. For instance, binary variables will follow a Bernoulli distribution, proportional variables follow a binomial distribution, and most count variables will follow a Poisson distribution.
If you have a very unusual variable that doesn’t follow one of these exponential family distributions, however, then a GLM will not be an appropriate choice. In other words, a GLM is not necessarily a magic fix!
11.2.2 Assumption 2: Correct link function
A closely-related assumption to assumption 1 above, is that we have chosen the correct link function for our model.
If we have done so, then there should be a linear relationship between our transformed model and our response variable; in other words, if we have chosen the right link function, then we have correctly “linearised” our model.
11.2.3 Assumption 3: Independence
We expect that the each observation or data point in our sample is independent of all the others. Specifically, we expect that our set of \(y\) response variables are independent of one another.
For this to be true, we have to make sure:
- that we aren’t treating technical replicates as true/biological replicates;
- that we don’t have observations/data points in our sample that are artificially similar to each other (compared to other data points);
- that we don’t have any nuisance/confounding variables that create “clusters” or hierarchy in our dataset;
- that we haven’t got repeated measures, i.e., multiple measurements/rows per individual in our sample
There is no diagnostic plot for assessing this assumption. To determine whether your data are independent, you need to understand your experimental design.
You might find this page useful if you’re looking for more information on what counts as truly independent data.
11.3 Other features to check
There is a handful of other features or qualities that can affect the quality of model fit, and therefore the quality of the inferences we draw from it.
These are not necessarily “formal” assumptions, but it’s good practice to check for them.
Lack of influential observations
A data point is overly influential, i.e., has high leverage, if removing that point from the dataset would cause large changes in the model coefficients. Data points with high leverage are typically those that don’t follow the same general “trend” as the rest of the data.
Lack of collinearity
Collinearity is when predictor variables are overly/strongly correlated with each other. This can make it very difficult to estimate the right beta coefficients and individual p-values for those predictors.
Dispersion
This is mentioned here for completeness, and as a bit of sizzle for next chapter when we talk about Poisson regression.
We won’t be worrying about dispersion in logistic regression specifically.
If you want more detail, you can skip ahead now to Section 12.6.
11.4 Assessing assumptions & quality
In linear modelling, we rely heavily on visuals to determine whether various assumptions were met.
When checking assumptions and assessing quality of fit for GLMs, we don’t use the panel of diagnostic plots that we used for linear models any more. However, there are some visualisations that can help us, plus several metrics we can calculate from our data or model.
| Is it a formal assumption? | How can I assess it? | |
|---|---|---|
Response variable comes from a distribution in the exponential family & The model uses the right link function |
Yes | Knowledge of the experimental design Some plots, e.g., posterior predictive check/uniform Q-Q, may help |
| Independent observations | Yes | Knowledge of experimental design There are no formal tests or plots that assess independence reliably |
| Influential observations | No | Cook’s distance/leverage plot |
| Lack of collinearity | No | Variance inflation factor |
| Dispersion | Sort of | Calculating the dispersion parameter Can also be visualised |
Let’s fit some useful diagnostic plots, using the diabetes and aphids example datasets.
diabetes <- read_csv("data/diabetes.csv")
glm_dia <- glm(test_result ~ glucose * diastolic,
family = "binomial",
data = diabetes)We’re going to rely heavily on the performance package in R for assessing the assumptions and fit of our model.
The check_model function will be our primary workhorse. This function automatically detects the type of model you give it, and produces the appropriate panel of plots all by itself. (So, so cool. And yes, it works for linear models too!)
check_model(glm_dia)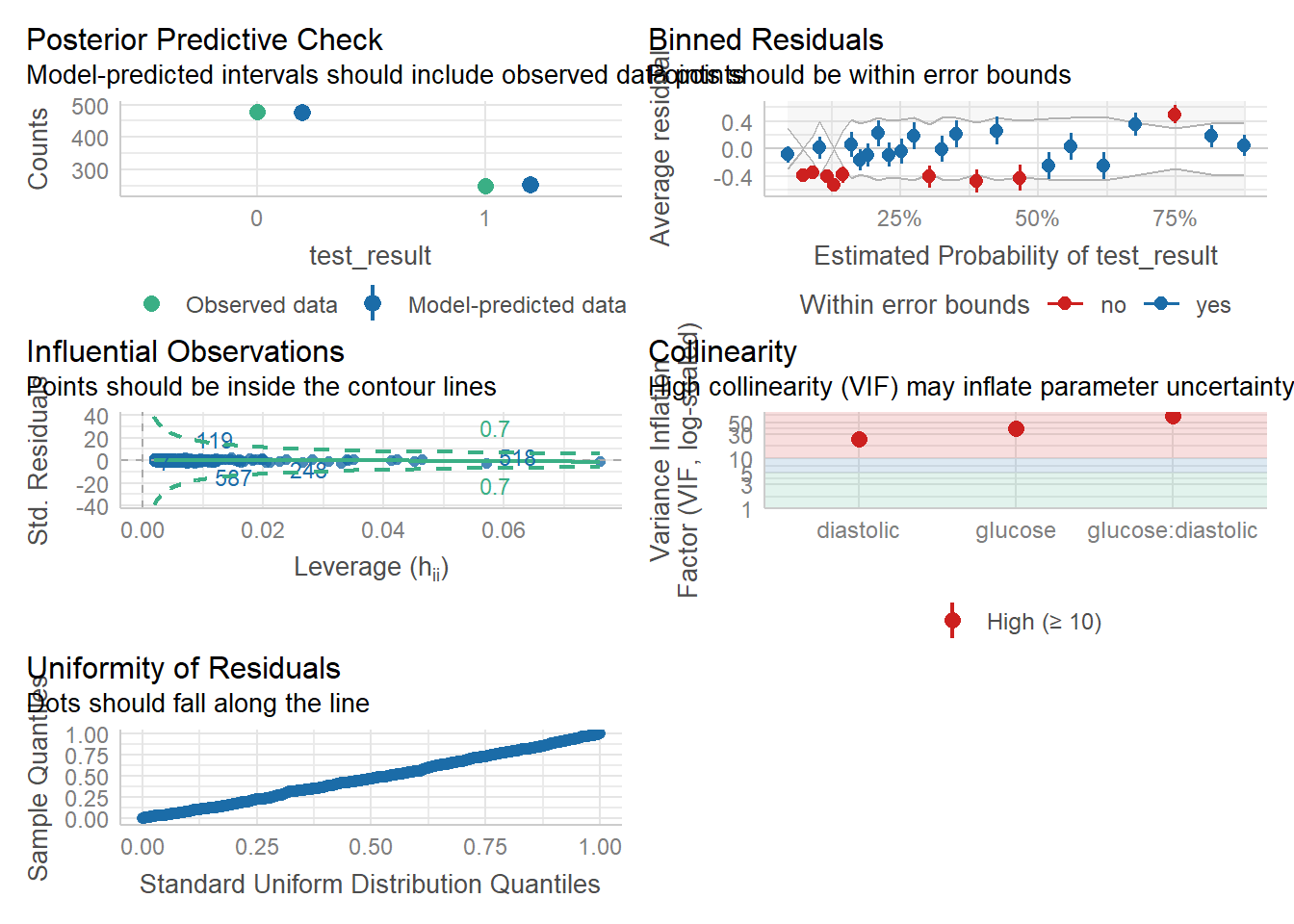
diabetes_py = pd.read_csv("data/diabetes.csv")
model = smf.glm(formula = "test_result ~ glucose * diastolic",
family = sm.families.Binomial(),
data = diabetes_py)
glm_dia_py = model.fit()11.4.1 Influential observations
We can check for outliers via a leverage plot (in performance), which you may remember from linear modelling.
Ideally, data points would fall inside the green contour lines. Data points that don’t will be highlighted in red.
check_model(glm_dia, check = 'outliers')Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.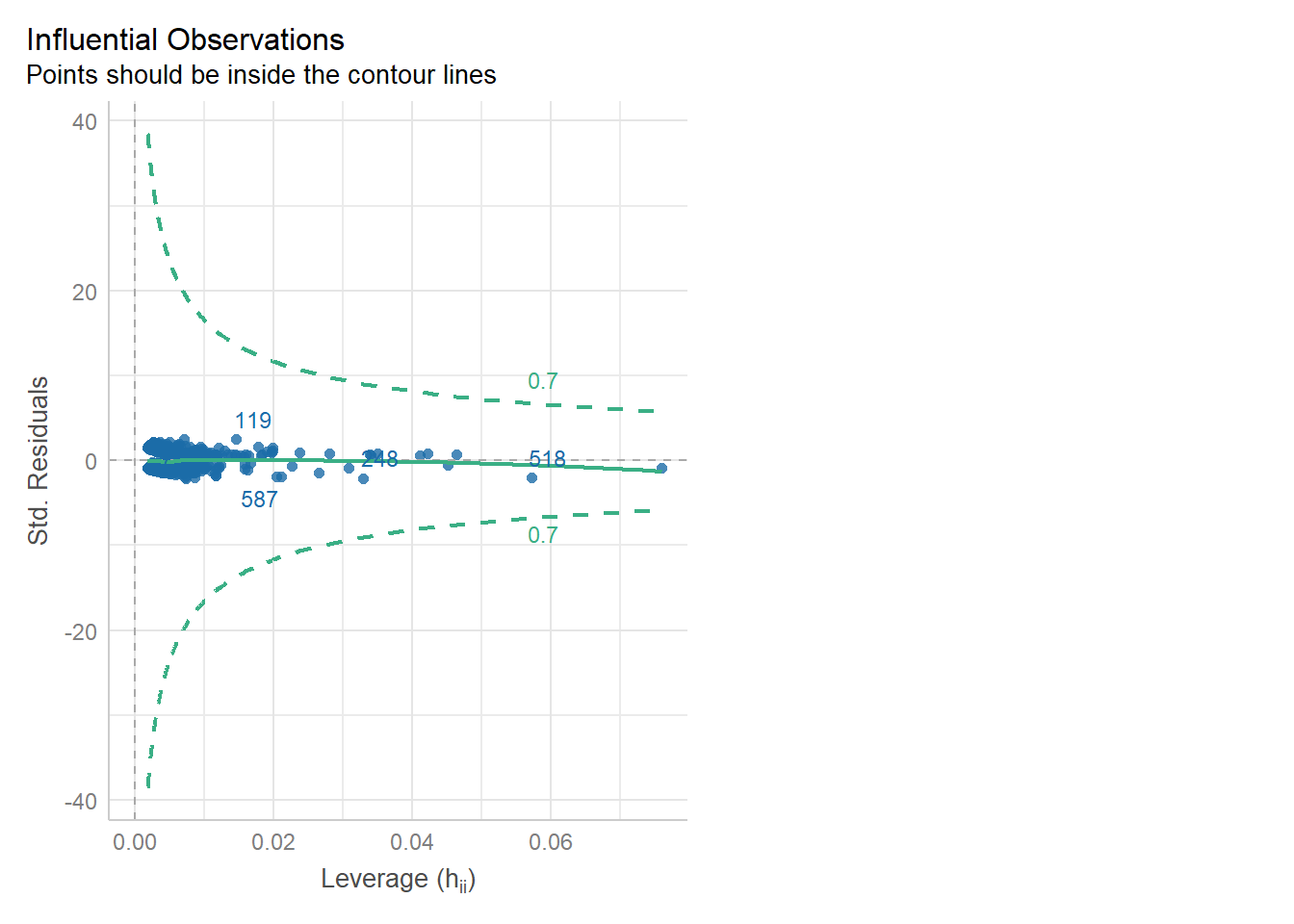
Alternatively, we can use the check_outliers function:
check_outliers(glm_dia, threshold = list('cook' = 0.5))OK: No outliers detected.
- Based on the following method and threshold: cook (0.5).
- For variable: (Whole model)We can use the get_influence function to extract information like leverage, Cook’s distance etc. about all of our data points:
influence = glm_dia_py.get_influence()Then, we can visualise and interrogate that information.
We can produce a Cook’s distance plot (this uses matplotlib):
cooks = influence.cooks_distance[0]
plt.stem(np.arange(len(cooks)), cooks, markerfmt=",")
plt.axhline(0.5, color='red', linestyle='--', label='Threshold')
plt.xlabel('Observation')
plt.ylabel("Cook's Distance")
plt.title("Cook's Distance Plot")
plt.legend()
plt.show()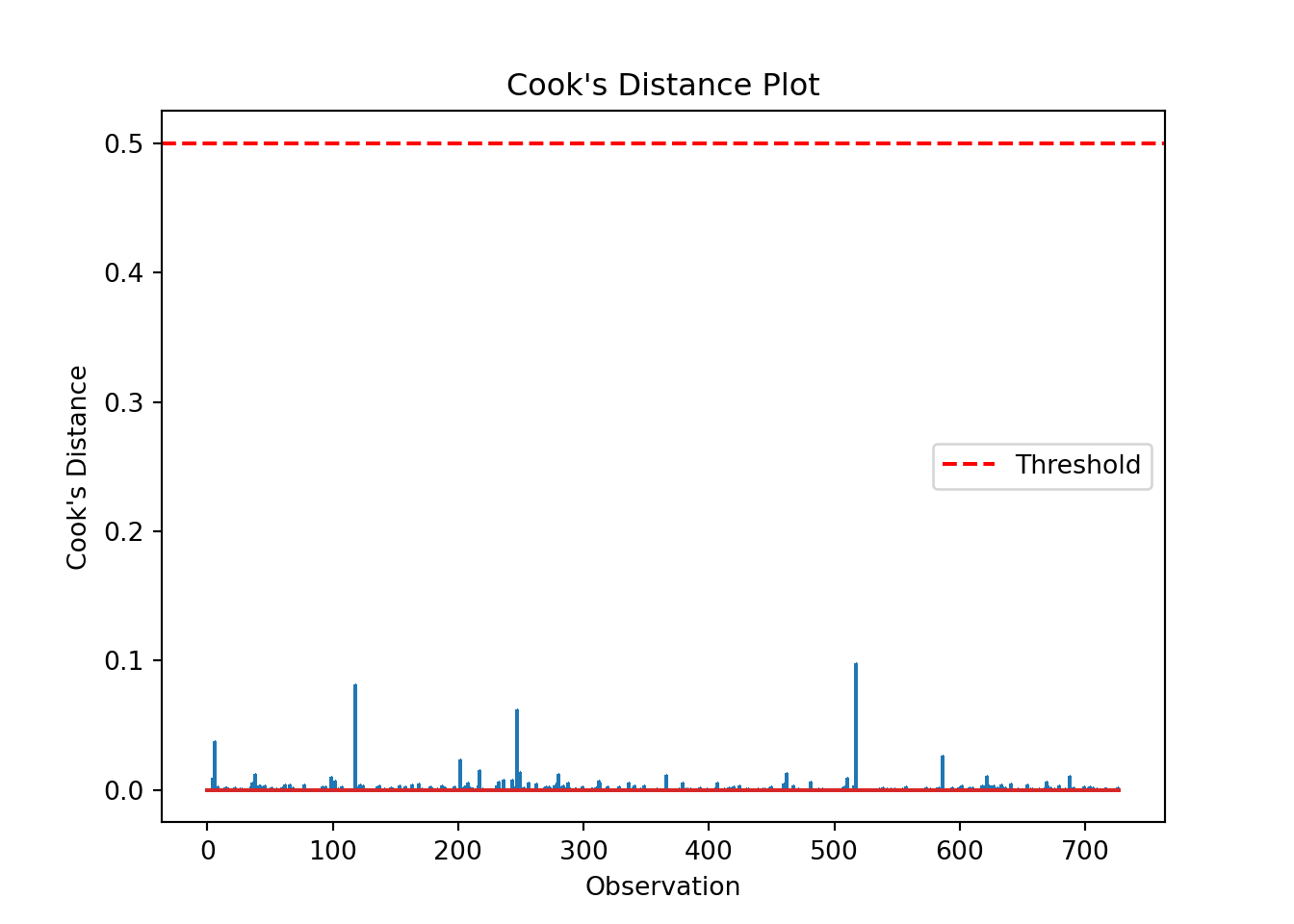
and/or we can extract a list of data points with a Cook’s distance greater than some specified threshold:
influential_points = np.where(cooks > 0.5)[0] # Set appropriate threshold here
print("Influential points:", influential_points)Influential points: []The list is empty, indicating no high leverage points that we need to worry about.
Dealing with “outliers”
Remember:
- Outliers and influential points aren’t necessarily the same thing; all outliers need to be followed up, to check if they are influential in a problematic way
- You can’t just drop data points because they are inconvenient! This is cherry-picking, and it can impact the quality and generalisability of your conclusions
11.4.2 Lack of collinearity
The best way to assess whether we have collinearity is to look at something called the variance inflation factor (VIF).
When calculating VIF for a model, a separate VIF value will be generated for each predictor. VIF >5 is worth an eyebrow raise; VIF >10 definitely needs some follow-up; and VIF >20 suggests a really strong correlation that is definitely problematic.
check_collinearity(glm_dia)Model has interaction terms. VIFs might be inflated.
Try to center the variables used for the interaction, or check
multicollinearity among predictors of a model without interaction terms.# Check for Multicollinearity
High Correlation
Term VIF VIF 95% CI Increased SE Tolerance Tolerance 95% CI
glucose 37.99 [32.97, 43.80] 6.16 0.03 [0.02, 0.03]
diastolic 24.24 [21.06, 27.92] 4.92 0.04 [0.04, 0.05]
glucose:diastolic 69.45 [60.21, 80.14] 8.33 0.01 [0.01, 0.02]We can also visualise the VIFs, if we prefer, with the VIF plot in check_model:
check_model(glm_dia, check = "vif")Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.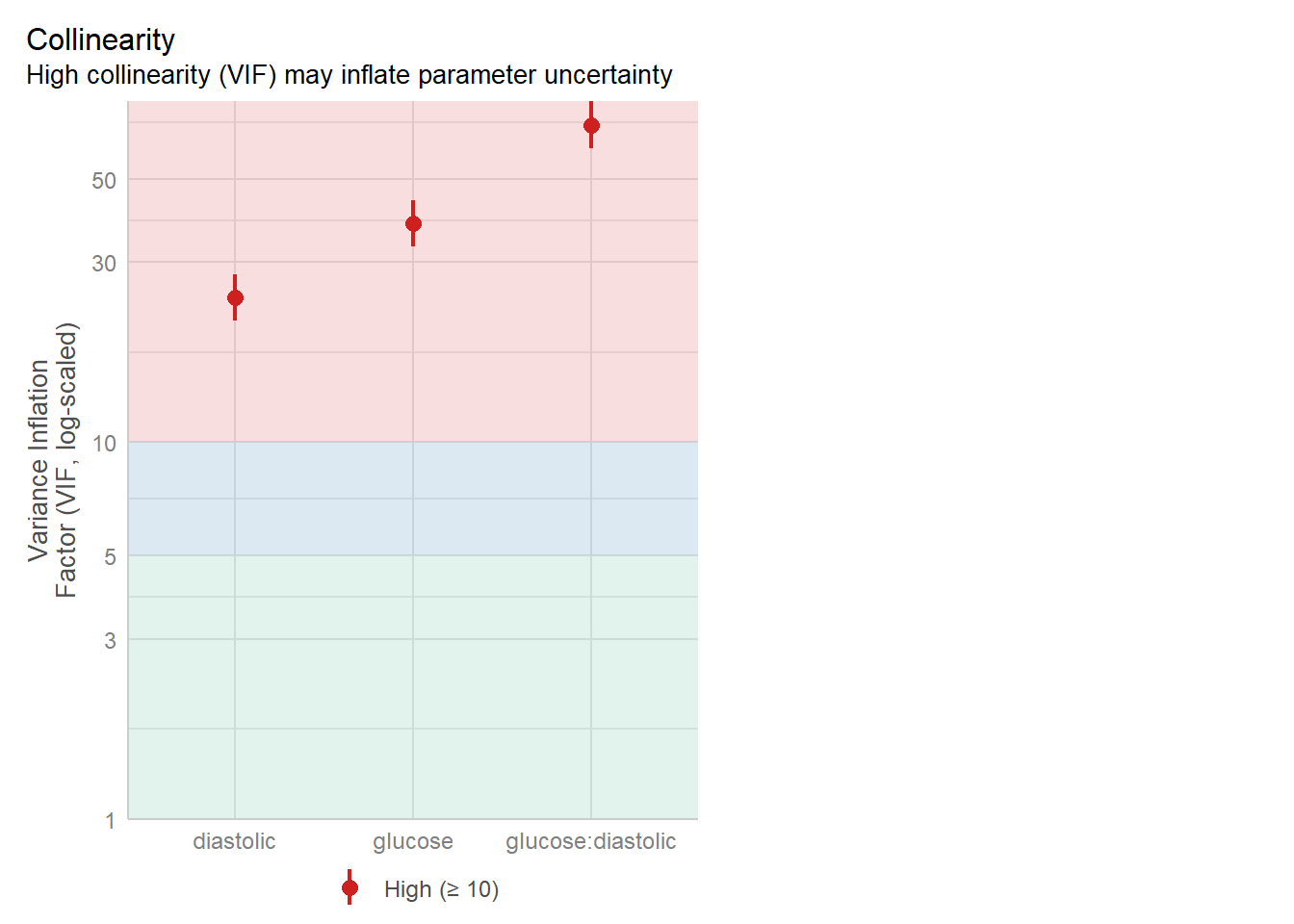
The statsmodels package contains a function for calculating VIF.
It uses the original dataset, rather than the model, to do this. This means you have to manually exclude the response variable, and then use the dmatrix function from patsy to .
To make sure all that
from statsmodels.stats.outliers_influence import variance_inflation_factor
from patsy import dmatrix
# Drop the response variable
X = diabetes_py.drop(columns='test_result')
# Create design matrix based on model formula
X = dmatrix("glucose * diastolic", data=diabetes_py, return_type='dataframe')
# Calculate VIF for each feature
vif_data = pd.DataFrame()
vif_data['feature'] = X.columns
vif_data['VIF'] = [variance_inflation_factor(X.values, i)
for i in range(X.shape[1])]
print(vif_data) feature VIF
0 Intercept 600.539179
1 glucose 36.879125
2 diastolic 17.407953
3 glucose:diastolic 63.987700There is definitely some collinearity going on in our model - these VIF values are way over 10.
The most likely culprit for this is the interaction term - let’s see if we can improve things by dropping it:
glm_dia_add <- glm(test_result ~ glucose + diastolic,
family = "binomial",
data = diabetes)check_collinearity(glm_dia_add)# Check for Multicollinearity
Low Correlation
Term VIF VIF 95% CI Increased SE Tolerance Tolerance 95% CI
glucose 1.02 [1.00, 1.78] 1.01 0.98 [0.56, 1.00]
diastolic 1.02 [1.00, 1.78] 1.01 0.98 [0.56, 1.00]check_model(glm_dia_add, check = "vif")Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.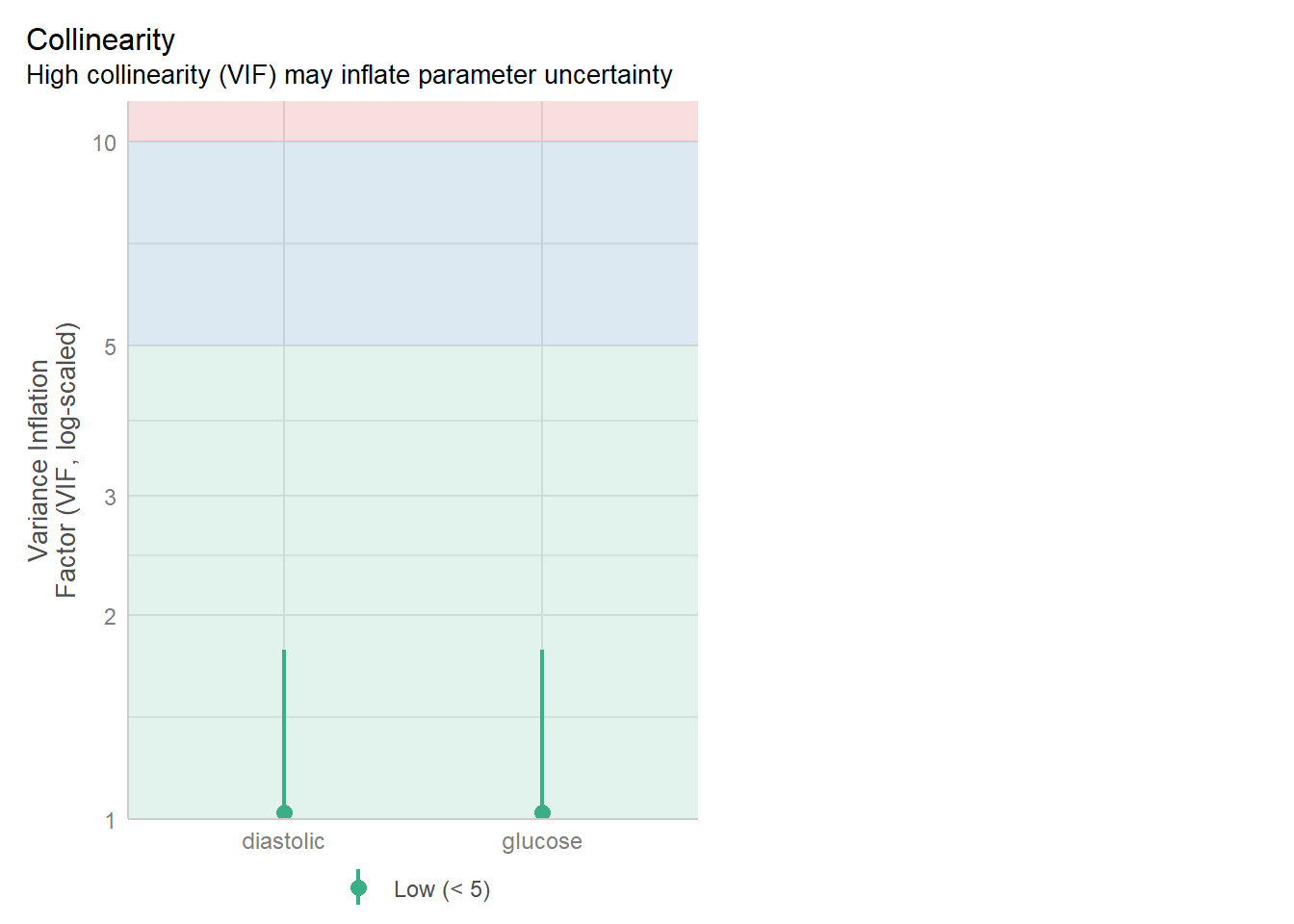
We will try again, this time without manually adding the interaction:
# 1. Drop the response variable
X = diabetes_py.drop(columns='test_result')
# 2. Add constant column for intercept
X = sm.add_constant(X)
# Calculate VIF for each feature
vif_data = pd.DataFrame()
vif_data['feature'] = X.columns
vif_data['VIF'] = [variance_inflation_factor(X.values, i)
for i in range(X.shape[1])]
print(vif_data) feature VIF
0 const 42.747425
1 glucose 1.052426
2 diastolic 1.052426Much better!
11.5 Exercises
11.5.1 Revisiting rats and levers (again)
Exercise
Level:
In Exercise 8.6.1 and Exercise 9.6.3, you worked through the levers dataset, fitting an appropriate model and then assessing its significance.
Now, using what you’ve learned in this chapter, assess whether this logistic regression is appropriate:
levers <- read_csv("data/levers.csv")
levers <- levers %>%
mutate(incorrect_presses = trials - correct_presses)
glm_lev <- glm(cbind(correct_presses, incorrect_presses) ~ stress_type * sex + rat_age,
family = binomial,
data = levers)levers = pd.read_csv("data/levers.csv")
levers['incorrect_presses'] = levers['trials'] - levers['correct_presses']
model = smf.glm(formula = "correct_presses + incorrect_presses ~ stress_type * sex + rat_age",
family = sm.families.Binomial(),
data = levers)
glm_lev = model.fit()
Answer
Consider the response variable
Is a logistic regression, with a logit link function, appropriate?
For the answer to be “yes”, then our response variable needs to be either a binary or a proportional variable (binomially distributed). We’re looking for those success/fail trials.
Sometimes, it can help to visualise the design:
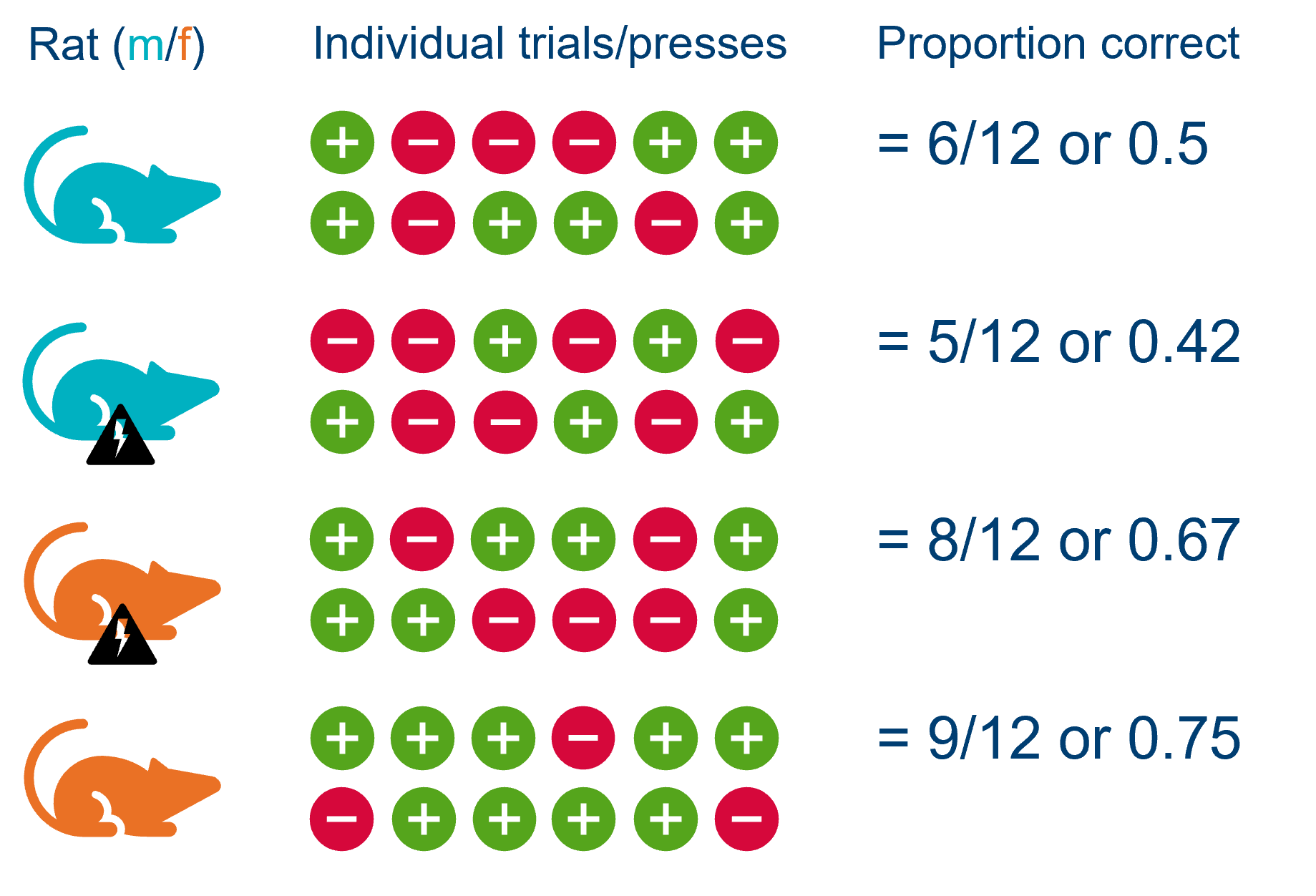
We can see that for each rat, the proportion score is made up of a series of trials, each of which can either be correct (+) or incorrect (-).
Consider independence
Again, we need to think about the design.
It might initially seem as if we have multiple observations per animal - since each rat pressed a whole bunch of levers - but actually if we look at each row of our dataset:
head(levers)# A tibble: 6 × 8
rat_id stress_type sex rat_age trials correct_presses prop_correct
<dbl> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
1 1 control female 13 20 7 0.35
2 2 control female 15 20 11 0.55
3 3 control female 11 20 5 0.25
4 4 control female 15 20 5 0.25
5 5 stressed female 13 20 8 0.4
6 6 stressed male 14 20 8 0.4
# ℹ 1 more variable: incorrect_presses <dbl>levers.head() rat_id stress_type sex ... correct_presses prop_correct incorrect_presses
0 1 control female ... 7 0.35 13
1 2 control female ... 11 0.55 9
2 3 control female ... 5 0.25 15
3 4 control female ... 5 0.25 15
4 5 stressed female ... 8 0.40 12
[5 rows x 8 columns]it becomes clear that each row of the dataset represents a rat, rather than each row representing a separate trial/button press.
In other words: by collecting the proportion of correct trials, we have averaged across the rat, and so it only appears once in our dataset.
This means that each of the rows of the dataset truly are independent.
Now, this does make some assumptions about the nature of the rats’ relationships to each other. Some non-independence could be introduced if:
- Some of the rats are genetically more similar to each other, e.g., if multiple rats from the same litter were included
- Rats were trained together before testing
- Rats were exposed to stress in one big batch (i.e., putting them all in one cage with the smell of a predator) rather than individually
Influential observations
check_model(glm_lev, check = 'outliers')Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.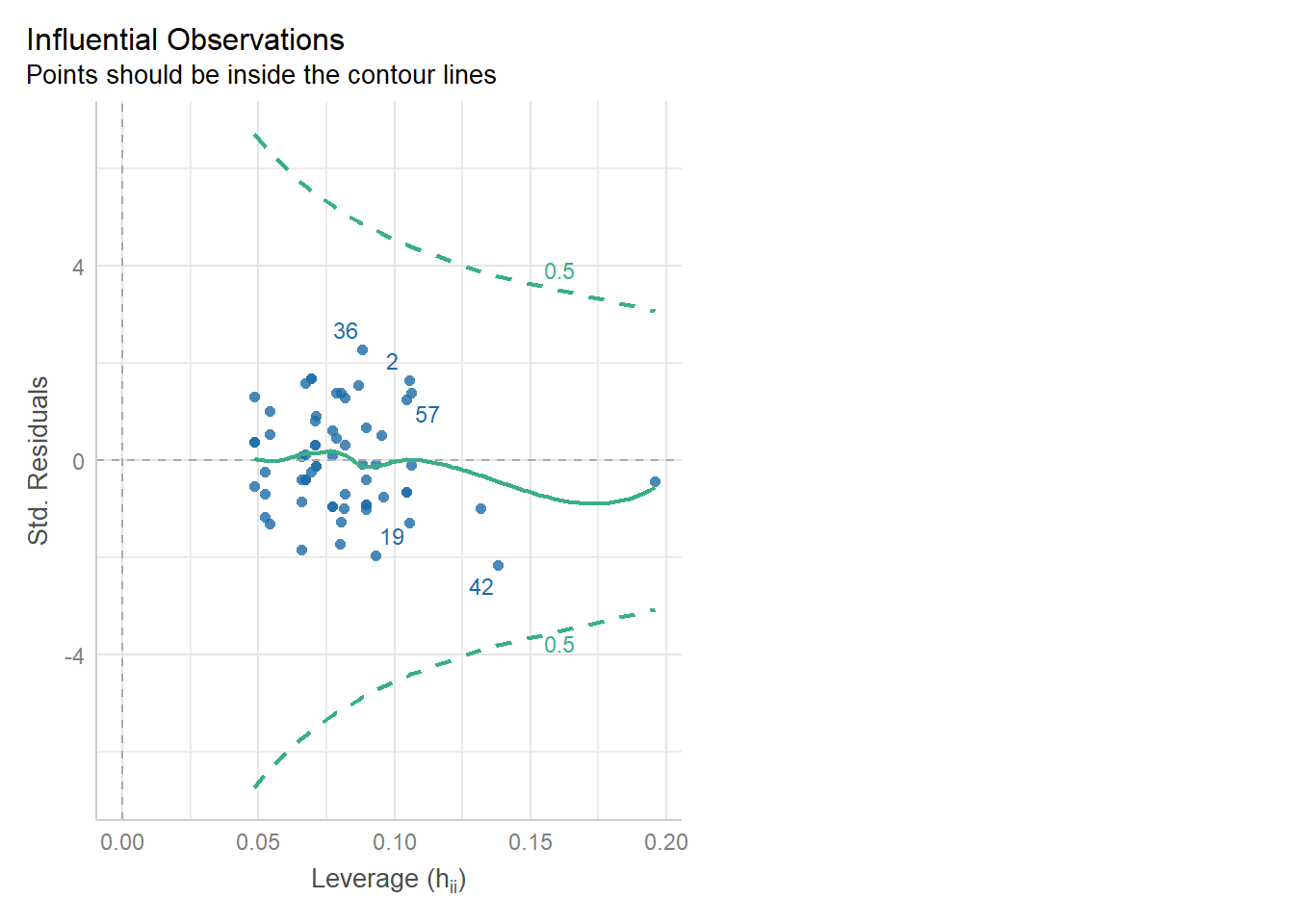
check_outliers(glm_lev)OK: No outliers detected.
- Based on the following method and threshold: cook (0.5).
- For variable: (Whole model)influence = glm_lev.get_influence()
cooks = influence.cooks_distance[0]
plt.stem(np.arange(len(cooks)), cooks, markerfmt=",")
plt.axhline(0.5, color='red', linestyle='--', label='Threshold')
plt.xlabel('Observation')
plt.ylabel("Cook's Distance")
plt.title("Cook's Distance Plot")
plt.legend()
plt.show()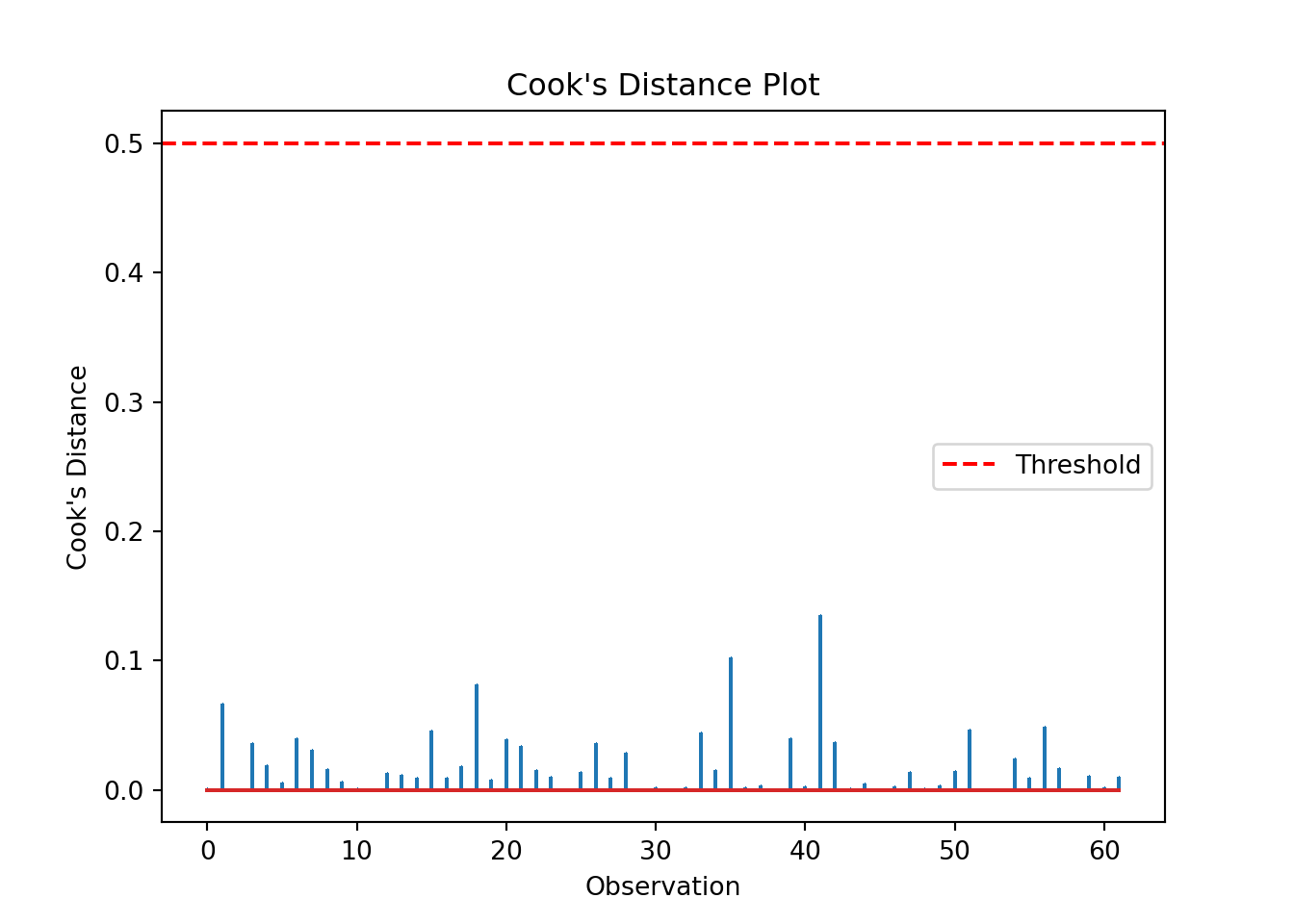
influential_points = np.where(cooks > 0.5)[0] # Set appropriate threshold here
print("Influential points:", influential_points)Influential points: []Looks good!
Collinearity
check_collinearity(glm_lev)# Check for Multicollinearity
Low Correlation
Term VIF VIF 95% CI Increased SE Tolerance Tolerance 95% CI
stress_type 1.81 [1.40, 2.64] 1.34 0.55 [0.38, 0.72]
sex 2.23 [1.66, 3.28] 1.49 0.45 [0.30, 0.60]
rat_age 1.02 [1.00, 287.92] 1.01 0.98 [0.00, 1.00]
stress_type:sex 2.70 [1.97, 4.01] 1.64 0.37 [0.25, 0.51]check_model(glm_lev, check = "vif")Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.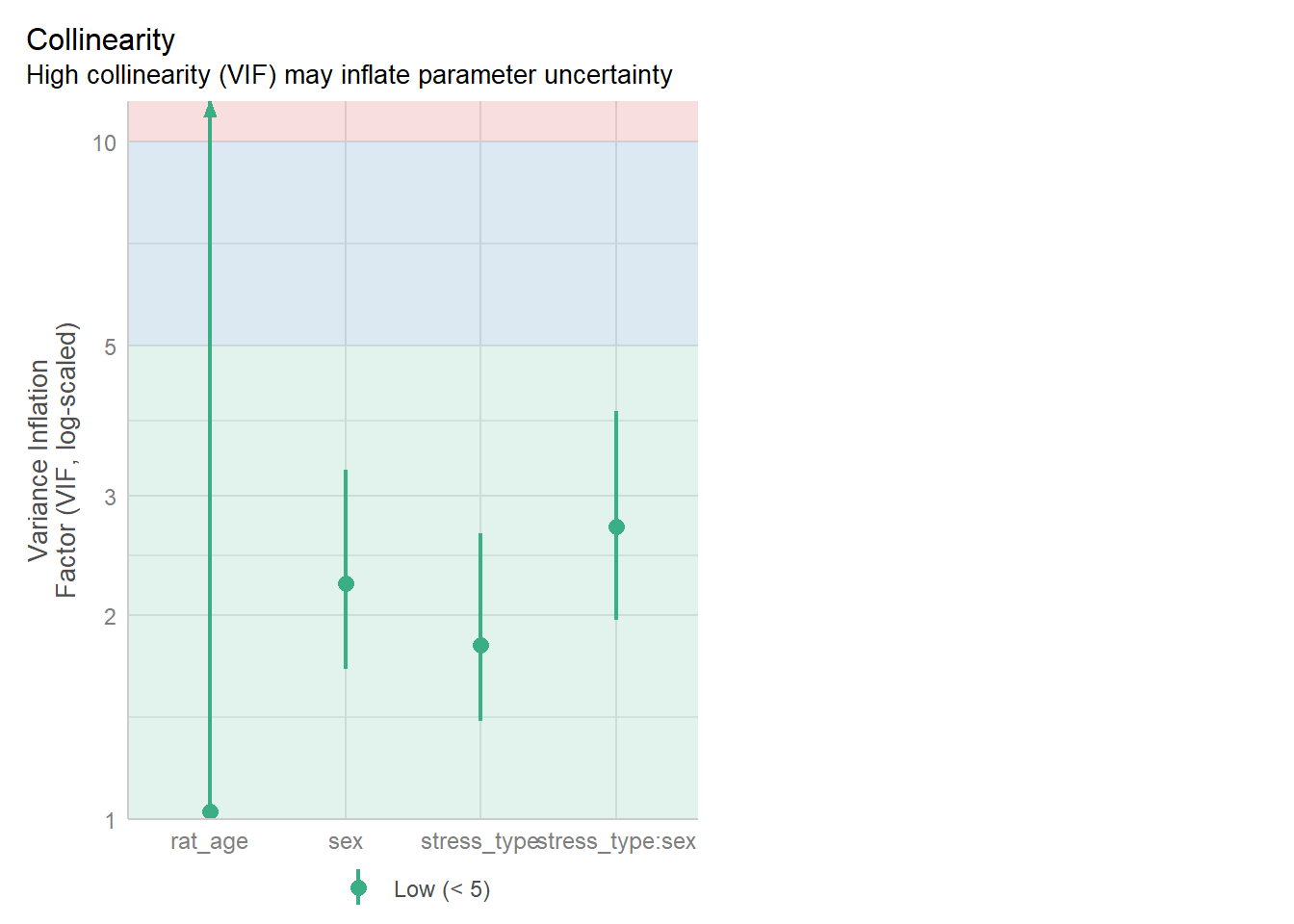
Remember to drop the response variables (and also rat ID in this case).
# Drop the response variable
X = levers.drop(columns=['rat_id','correct_presses','prop_correct','incorrect_presses'])
# Create design matrix
X = dmatrix("stress_type * sex + rat_age", data=levers, return_type='dataframe')
# Calculate VIF for each feature
vif_data = pd.DataFrame()
vif_data['feature'] = X.columns
vif_data['VIF'] = [variance_inflation_factor(X.values, i)
for i in range(X.shape[1])]
print(vif_data) feature VIF
0 Intercept 48.157694
1 stress_type[T.stressed] 1.768004
2 sex[T.male] 2.190167
3 stress_type[T.stressed]:sex[T.male] 2.799690
4 rat_age 1.016442Again, all good - none of our predictors have a concerning VIF.
Collectively, it would seem that our model does indeed meet the assumptions, and there are no glaring obstacles to us proceeding with a significance test and an interpretation.
11.5.2 Seed germination
Level:
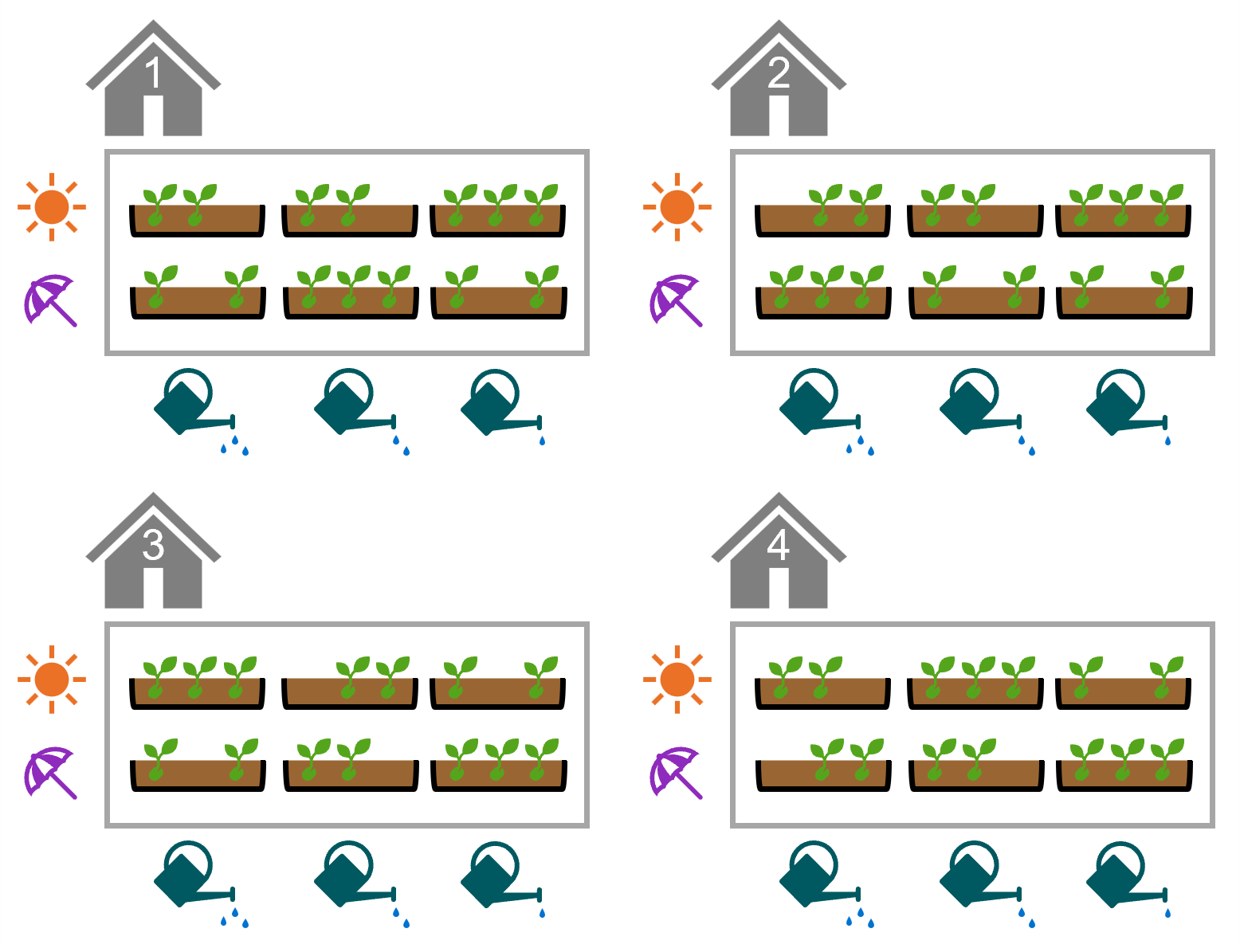
This exercise uses a new dataset, seeds, which is all about a seed germination experiment.
Each row of the dataset represents a seed tray, in which 25 seeds were planted. The trays were treated with one of two light conditions (sun, shade) and one of three watering frequencies (low, medium, high).
The researchers want to know whether either or both of these predictors have an affect on the proportion of seeds that successfully germinate.
In this exercise, you should:
- Visualise the data
- Fit a suitable model
- Test the assumptions and quality of model fit
- Decide whether to draw a biological conclusion from the data
seeds <- read_csv("data/seeds.csv")seeds = pd.read_csv("data/seeds.csv")There is no formal worked answer provided. However, all of the code you will need can be adapted from the diabetes and aphids examples worked through in the chapter.
You are also strongly encouraged to work with, or share your answer with, a neighbour.
Hints
- Consider a possible interaction effect
- Look closely at the dataset itself, and all its columns
- You should find at least two issues and/or failures of assumptions!
It may help to visualise the experimental design (the number of trays has been simplified so that the figure fits on the page):
11.6 Summary
While generalised linear models make fewer assumptions than standard linear models, we do still expect certain things to be true about the model and our variables for GLMs to be valid.
Key points
- For a generalised linear model, we assume that we have chosen the correct link function, that our response variable follows a distribution from the exponential family, and that our data are independent
- We also want to check that there are no overly influential points, no collinearity, and that the dispersion parameter is close to 1
- To assess some of these assumptions/qualities, we have to rely on our understanding of our dataset
- For others, we can calculate metrics like Cook’s distance and variance inflation factor, or produce diagnostic plots