Customise shell scripts to work with inputs defined by the user.
Define Bash variables based on commands.
8.1 Custom Inputs
When we discussed Shell scripts, we wrote a script that counted the number of atoms on a specific PDB file (in our example cubane.pdb). But what if we wanted to give it as input a file of our choice? We can make our script more versatile by using a special shell variable that means “the first argument on the command line”. Here is our new script, modified from the previous section:
#!/bin/bash# print a message to the userecho"Processing file: $1"# count the number of lines containing the word "ATOM"cat"$1"|grep"ATOM"|wc-l
The main change in our script is that we used a special variable called $1 to indicate the file that we want to process will be given by the user from the command line. This variable means “the first argument passed to the shell script”. You can use any number of these, for example $2 would mean “the second argument passed to the shell script”. These are known as positional argument variables.
So, if we run our modified script, this is the result:
bash count_atoms.sh ethane.pdb
Processing file: ethane.pdb
8
This is a much more flexible script, as the input can now be specified by the user.
8.2 Bash Variables
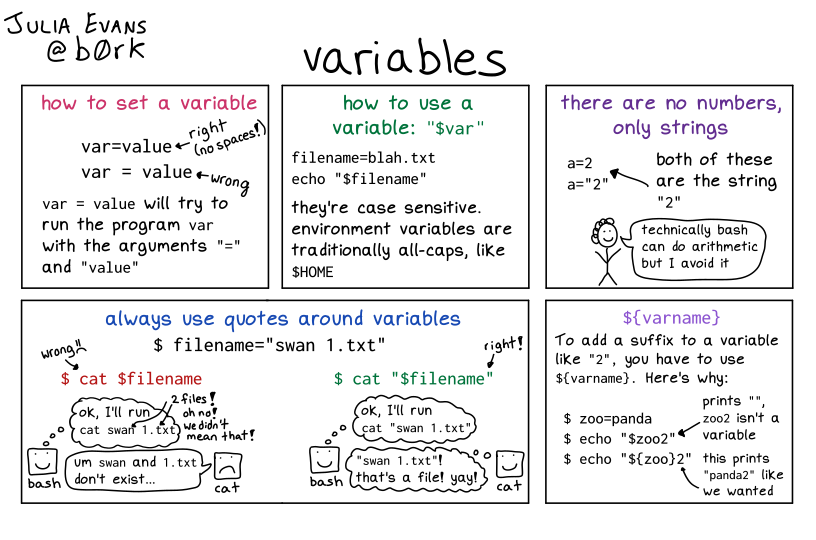
Variables in Bash always start with the $ symbol. We have already seen the special variables called $1, $2 (which take input from the user). However, we can also create variables ourselves, with the following syntax:
molecule="ethane"
This would create a variable named “molecule” containing the text “ethane”. Notice that there should be no space between the variable name (“molecule”) and its value (“ethane”).
Once we create a variable, we need to prefix it with $ every time we want to use it. For example, to see the value stored inside a variable we can use the echo command:
echo"$molecule"
ethane
Single ' or double " quotes?
Wrapping variables in single or double quotes makes a difference.
When you use double quotes the shell will interpret the values in the variable, as shown in the example above. However, if you use single quotes, the shell will not interpret those variable values, instead printing the text as is:
echo'My molecule is: $molecule'
My molecule is $molecule
In this case, our variable is storing the name of one of our molecules, so we could use it to look at the content of our file:
grep"ATOM""${molecule}.pdb"
ATOM 1 C 1 -0.752 0.001 -0.141 1.00 0.00
ATOM 2 C 1 0.752 -0.001 0.141 1.00 0.00
ATOM 3 H 1 -1.158 0.991 0.070 1.00 0.00
ATOM 4 H 1 -1.240 -0.737 0.496 1.00 0.00
ATOM 5 H 1 -0.924 -0.249 -1.188 1.00 0.00
ATOM 6 H 1 1.158 -0.991 -0.070 1.00 0.00
ATOM 7 H 1 0.924 0.249 1.188 1.00 0.00
ATOM 8 H 1 1.240 0.737 -0.496 1.00 0.00
One thing to note here is that we included the variable name within {}. The reason is that this allows us to combine the value of a variable with other text.
Take these two examples:
echo"$molecule_copy"echo"${molecule}_copy"
The first command would give us an empty output because Bash would think there is a variable called “molecule_copy”, but such a variable does not exist (and by default the shell assumes its value is empty). In the second command, because we included the variable name in {}, then this is not a problem and the output we get is “ethane_copy”.
In conclusion: always include {} when using your variables in scripts. It is also good practice to always include variables within double " quotes. The reasons are more subtle, but see this StackOverflow post to learn more about it.
8.2.1 Environment Variables
There are many default variables that are automatically created when we open the terminal. These are called environment variables, which as a convention are always named with upppercase. For example the variable $HOME stores the user’s home directory.
Try running:
echo$HOME
8.2.2 Variables and Commands
Very often we may want to create a variable with the result of evaluating a command. The syntax to do this is:
variable=$(command)
For example, let’s say we wanted to create a variable that stores the results of the grep command we ran earlier:
ethane_atoms=$(cat ethane.pdb |grep"ATOM"|wc-l)
Running this command generates no output. Instead the output of the command is stored inside our variable. We can print the content of the variable with:
echo"$ethane_atoms"
8
8.3 Exercises
Exercise 1 - Variable comprehension
Level:
(Note: this is a conceptual exercise, you don’t need to use your own terminal.)
Assuming that we have a user named robin, what would be the output of the echo command below?
No, this would be the output of echo "${datadir}".
No, this would be the output of echo "${datadir}/molecules".
Yes, this is the correct answer. It is the equivalent of running the command ls /home/robin/Desktop/data-shell/molecules/.
No, the full filepath is not included in the output.
Exercise 2 - Positional arguments
Level:
Take the following code, which counts the number of carbon atoms in one of our molecule files:
# print a messageecho"The number of C atoms in ethane.pdb is:"# count carbon "C" atomscat ethane.pdb |grep"ATOM"|grep"C"|wc-l
We want to generalise this code, such that it works on different molecule input files and to search for different types of atoms.
Using nano create a new script called count_atom_type.sh and copy-paste the code above.
Test that the script works by running it with bash count_atom_type.sh.
Modify the script to take two inputs provided by the user:
the first input is the filename to process
the second input is the type of atom to search for
Use your modified script to count the number of hydrogen atoms (“H”) in one of the molecule files.
Hint
Recall that the special variables $1, $2, etc., can be used to store, respectively, the first, second, etc., user-provided arguments to the script.
Here is an example of the command (and its output) that you should be able to run with your final script:
bash count_atom_type.sh methane.pdb H
The number of H atoms in methane.pdb is:
4
Answer
In our modified script we use the $1 and $2 variables to capture input given by the user.
Here is our script, which generalises the code given:
# print a messageecho"The number of $2 atoms in $1 is:"# count carbon "C" atomscat"$1"|grep"ATOM"|grep"$2"|wc-l
With this code saved in a script called count_atom_type.sh, we should be able to run it like this, for example:
bash count_atom_type.sh octane.pdb H
The number of H atoms in octane.pdb is:
18
Exercise 3 - Variables and commands
Level:
Let’s continue working on our earlier script count_atoms.sh. Let’s say that instead of printing the number of atoms to the console, we would want the result to be saved in a file named as <MOLECULE>_atoms.txt (where <MOLECULE> is the name of the respective molecule file).
To achieve this, we will use the help of a new command called basename. This command returns the filename in a path. For example:
basename molecules/ethane.pdb
ethane.pdb
Furthermore, we can also add some text we want removed at the end of the filename (usually used to remove the file extension). For example:
basename molecules/ethane.pdb ".pdb"
ethane
Based on this knowledge, modify the count_atoms.sh script to save the output into a file with the name and extension as mentioned above. For example, this command:
count_atoms.sh ethane.pdb
Should create a new file called ethane_atoms.txt
Hint
Inside the script, create a variable molecule that stores the basename of the user-provided input file.
Remember that user-provided inputs are stored in the special variables $1, $2, etc.
Recall that you can store the result of a command in a variable, using the syntax: variable=$(command)
Answer
Here is our modified count_atoms.sh script:
#!/bin/bash# store the molecule name in a new variable# remove the ".pdb" extension from the namemolecule=$(basename$1".pdb")# print a message to the userecho"Processing file: $1"# count the number of lines containing the word "ATOM"# save the output to a new filecat"$1"|grep"ATOM"|wc-l>"${molecule}_atoms.txt"
Running this script now creates a file with the name of the molecule, instead of printing the number of atoms to the console.
Exercise 4 - Positional arguments (advanced)
Level:
Go back to the data-shell folder for this exercise.
Write a shell script called longest.sh that takes two inputs: the name of a directory and a file extension.
The script should then return the name of the file with the most lines in that directory with that extension. For example:
bash longest.sh molecules pdb
Should print the name of the .pdb file in the molecules folder that has the most lines.
Using your script determine what is the longest PDB file in molecules and the longest CSV file in coronavirus/variants.
Hint
Before writing the script, first test how you would achieve this for a directory and file extension of your choice.
Once that is working, you can then try to generalise your script to take user inputs (using $1 and $2 special variables).
The commands you can use to help you are wc -l (to count the number of lines in a file), sort -n (to sort the input numerically), head and tail combined to get a specific line from an input.
Remember that to get a specific line from an input, you can use the trick head -n <LINE NUMBER YOU WANT> | tail -n 1.
Answer
Here is a script that would do what is requested:
#!/bin/bash# This script takes two arguments:# 1. a directory name# 2. a file extension# and prints the name of the file in that directory # with the most lines which matches the file extensionwc-l$1/*.$2|sort-n-r|head-n 2 |tail-n 1
Here is an explanation of each step of our chain of commands:
With wc -l $1/*.$2 we:
count lines in input files with wc -l
we specify the input files to this command using $1/*.$2, where $1 is the name of the directory given by the user and *.$2 is used to match all file names (*) with a given extension specified by the user ($2)
With sort -n -r we get the output of the previous command sorted in numeric and reverse order (so we have largest files first)
With head -n 2 | tail -n 1 we get the 2nd line comming out of the previous command. The reason we do this is that the wc command also counts the total number of lines across all files, which is not what we want. Therefore we get the 2nd line of the output, which corresponds to the file with the most lines.
We could then run this script on both of those directories:
bash longest.sh molecules pdb
30 molecules/octane.pdb
And:
bash longest.sh coronavirus/variants csv
256 coronavirus/variants/all_countries.csv
Here is a diagram illustrating what is happening at each step of our chain of commands (using the molecules pdb files as an example):
Positional variables such as $1, $2, $3, etc., can be used to store input values specified by the user when running the script.
Environment variables are default variables created by the shell. For example $HOME stores the user’s home directory path. These are conventionally named with uppercase.
Custom variables can be defined with the syntax:
variable="value" if we want the variable to contain a fixed value.
variable=$(command) if we want the variable to contain the result of running a command.
The value stored in a variable can be printed using echo "$variable".
Variable names should be wrapped in {} if concatenating with other text. For example echo ${variable}_suffix will add “suffix” to the value stored in the variable.