5 Text Manipulation
- Inspect the content of text files (
head,tail,cat,zcat,less). - Use the
*wildcard to work with multiple files at once. - Redirect the output of a command to a file (
>,>>). - Find a pattern in a text file (
grep).
5.1 Looking Inside Files
Often we want to investigate the content of a file, without having to open it in a text editor. This is especially useful if the file is very large (as is often the case in bioinformatic applications).
For example, let’s take a look at the cubane.pdb file in the molecules directory. We will start by printing the whole content of the file with the cat command, which stands for “concatenate” (we will see why it’s called this way in a little while):
cd molecules
cat cubane.pdbCOMPND CUBANE
AUTHOR DAVE WOODCOCK 95 12 06
ATOM 1 C 1 0.789 -0.852 0.504 1.00 0.00
ATOM 2 C 1 -0.161 -1.104 -0.624 1.00 0.00
ATOM 3 C 1 -1.262 -0.440 0.160 1.00 0.00
ATOM 4 C 1 -0.289 -0.202 1.284 1.00 0.00
ATOM 5 C 1 1.203 0.513 -0.094 1.00 0.00
ATOM 6 C 1 0.099 1.184 0.694 1.00 0.00
ATOM 7 C 1 -0.885 0.959 -0.460 1.00 0.00
ATOM 8 C 1 0.236 0.283 -1.269 1.00 0.00
ATOM 9 H 1 1.410 -1.631 0.942 1.00 0.00
ATOM 10 H 1 -0.262 -2.112 -1.024 1.00 0.00
ATOM 11 H 1 -2.224 -0.925 0.328 1.00 0.00
ATOM 12 H 1 -0.468 -0.501 2.315 1.00 0.00
ATOM 13 H 1 2.224 0.892 -0.134 1.00 0.00
ATOM 14 H 1 0.240 2.112 1.251 1.00 0.00
ATOM 15 H 1 -1.565 1.730 -0.831 1.00 0.00
ATOM 16 H 1 0.472 0.494 -2.315 1.00 0.00
TER 17 1
ENDSometimes it is useful to look only at only the top few lines of a file (especially for very large files). We can do this with the head command:
head cubane.pdbCOMPND CUBANE
AUTHOR DAVE WOODCOCK 95 12 06
ATOM 1 C 1 0.789 -0.852 0.504 1.00 0.00
ATOM 2 C 1 -0.161 -1.104 -0.624 1.00 0.00
ATOM 3 C 1 -1.262 -0.440 0.160 1.00 0.00
ATOM 4 C 1 -0.289 -0.202 1.284 1.00 0.00
ATOM 5 C 1 1.203 0.513 -0.094 1.00 0.00
ATOM 6 C 1 0.099 1.184 0.694 1.00 0.00
ATOM 7 C 1 -0.885 0.959 -0.460 1.00 0.00
ATOM 8 C 1 0.236 0.283 -1.269 1.00 0.00By default, head prints the first 10 lines of the file. We can change this using the -n option, followed by a number, for example:
head -n 2 cubane.pdbCOMPND CUBANE
AUTHOR DAVE WOODCOCK 95 12 06Similarly, we can look at the bottom few lines of a file with the tail command:
tail -n 2 cubane.pdbTER 17 1
ENDFinally, if we want to open the file and browse through it, we can use the less command:
less cubane.pdbless will open the file and you can use ↑ and ↓ to move line-by-line or the Page Up and Page Down keys to move page-by-page. You can exit less by pressing Q (for “quit”). This will bring you back to the console.
less
When you open a file with the less program, you can also search for text within the file. To do this, press / and you will notice the bottom of the terminal changes to /. Now, type the word (or part of a word) that you want to search for and press Enter ↵.
Less will search of the word in the file and highlight it for you. If you want to move to the next match press n and to move to the previous match press Shift + n.
5.2 Count Words/Lines/Characters
Often it can be useful to count how many lines, words and characters a file has. We can use the wc command for this:
wc *.pdb 20 156 1158 cubane.pdb
12 84 622 ethane.pdb
9 57 422 methane.pdb
30 246 1828 octane.pdb
21 165 1226 pentane.pdb
15 111 825 propane.pdb
107 819 6081 totalIn this case, we used the * wildcard to count lines, words and characters (in that order, left-to-right) of all our PDB files. Often, we only want to count one of these things, and wc has options for all of them:
-lcounts lines only.-wcounts words only.-ccounts characters only.
For example, the following counts only the number of lines in each file:
wc -l *.pdb 20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 total5.3 Combining several files
Earlier, we said that the cat command stands for “concatenate”. This is because this command can be used to concatenate (combine) several files together. For example, if we wanted to combine all PDB files into one:
cat *.pdb5.4 Redirecting Output
The commands we’ve been using so far, print their output to the terminal. But what if we wanted to save it into a file? We can achieve this by redirecting the output of the command to a file using the > operator.
wc -l *.pdb > number_lines.txtNow, the output is not printed to the console, but instead sent to a new file. We can check that the file was created with ls.
If we use > and the output file already exists, its content will be replaced. If what we want to do is append the result of the command to the existing file, we can use >> instead. Let’s see this in practice in the next exercise.
5.5 Finding Patterns
Something it can be very useful to find lines of a file that match a particular text pattern. We can use the tool grep (“global regular expression print”) to achieve this.
Going back to our molecules directory (cd ../molecules), let’s find the word “ATOM” in our cubane.pdb molecule file:
grep "ATOM" cubane.pdbATOM 1 C 1 0.789 -0.852 0.504 1.00 0.00
ATOM 2 C 1 -0.161 -1.104 -0.624 1.00 0.00
ATOM 3 C 1 -1.262 -0.440 0.160 1.00 0.00
ATOM 4 C 1 -0.289 -0.202 1.284 1.00 0.00
ATOM 5 C 1 1.203 0.513 -0.094 1.00 0.00
ATOM 6 C 1 0.099 1.184 0.694 1.00 0.00
ATOM 7 C 1 -0.885 0.959 -0.460 1.00 0.00
ATOM 8 C 1 0.236 0.283 -1.269 1.00 0.00
ATOM 9 H 1 1.410 -1.631 0.942 1.00 0.00
ATOM 10 H 1 -0.262 -2.112 -1.024 1.00 0.00
ATOM 11 H 1 -2.224 -0.925 0.328 1.00 0.00
ATOM 12 H 1 -0.468 -0.501 2.315 1.00 0.00
ATOM 13 H 1 2.224 0.892 -0.134 1.00 0.00
ATOM 14 H 1 0.240 2.112 1.251 1.00 0.00
ATOM 15 H 1 -1.565 1.730 -0.831 1.00 0.00
ATOM 16 H 1 0.472 0.494 -2.315 1.00 0.00We can see the result is all the lines that matched this word pattern.
grep has many other options available, which can be useful depending on the result you want to get. Some of the more useful ones are illustrated below.
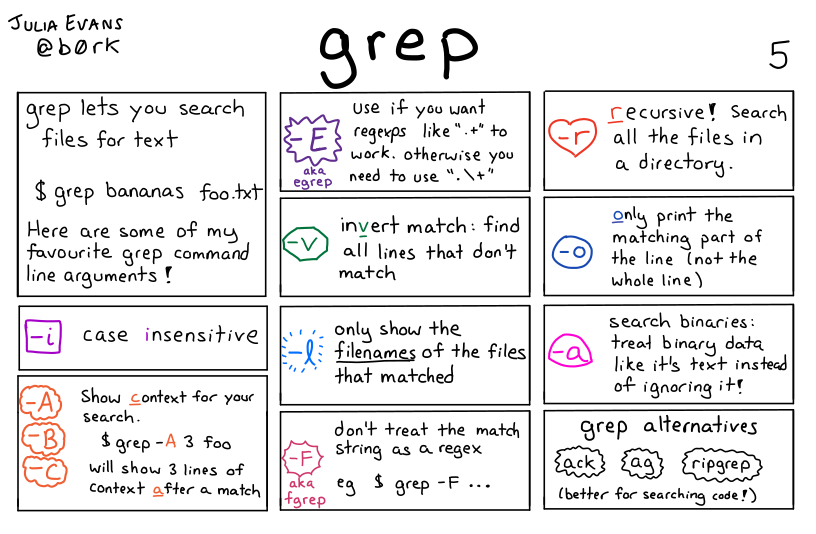
grep command by Julia Evans5.6 Exercises
5.7 Summary
- The
headandtailcommands can be used to look at the top or bottom of a file, respectively. - The
lesscommand can be used to interactively investigate the content of a file. Use ↑ and ↓ to browse the file and Q to quit and return to the console. - The
catcommand can be used to combine multiple files together. Thezcatcommand can be used instead if the files are compressed. - The
>operator redirects the output of a command into a file. If the file already exists, it’s content will be overwritten. - The
>>operator also redictects the output of a command into a file, but appends it to any content that already exists. - The
grepcommand can be used to find the lines in a text file that match a text pattern.