Construct command pipelines with two or more stages.
6.1 The | Pipe
In the previous section we ended with an exercise where we counted the number of lines matching the word “Alpha” in several CSV files containing variant classification of coronavirus virus samples from several countries.
We achieved this in three steps:
Combine all CSV files into one: cat *_variants.csv > all_countries.csv.
Create a new file containing only the lines that match our pattern: grep "Alpha" all_countries.csv > alpha.csv
Count the number of lines in this new file: wc -l alpha.csv
But what if we now wanted to search for a different pattern, for example “Delta”? It seems unpractical to keep creating new files every time we want to ask such a question from our data.
This is where one of the shell’s most powerful feature becomes handy: the ease with which it lets us combine existing programs in new ways.
The way we can combine commands together is using a pipe, which uses the special operator |. Here is our example using a pipe:
cat*_variants.csv |grep"Alpha"|wc-l
Notice how we now don’t specify an input to either grep nor wc. The input is streamed automatically from one tool to another through the pipe. So, the output of cat is sent to grep and the output from grep is then sent to wc.
6.2 Cut, Sort, Unique & Count
Let’s now explore a few more useful commands to manipulate text that can be combined to quickly answer useful questions about our data.
Let’s start with the command cut, which is used to extract sections from each line of its input. For example, let’s say we wanted to retrieve only the second field (or column) of our CSV file, which contains the clade classification of each of our omicron samples:
-d defines the delimiter used to separate different parts of the line. Because this is a CSV file, we use the comma as our delimiter. The tab is used as the default delimiter.
-f defines the field or part of the line we want to extract. In our case, we want the second field (or column) of our CSV file. It’s worth knowing that you can specify more than one field, so for example if you had a CSV file with more columns and wanted columns 3 and 7 you could set -f 3,7.
The next command we will explore is called sort, which sorts the lines of its input alphabetically (default) or numerically (if using the -n option). Let’s combine it with our previous command to see the result:
You can see that the output is now sorted alphabetically.
The sort command is often used in conjunction with another command: uniq. This command returns the unique lines in its input. Importantly, it only works as intended if the input is sorted. That’s why it’s often used together with sort.
Let’s see it in action, by continuing building our command:
We can see that now the output is de-duplicated, so only unique values are returned. And so, with a few simple commands, we’ve answered a very useful question from our data: what are the unique variants in our collection of samples?
Alphabetic or numeric sort?
Note that, by default the sort command will order input lines alphabetically. So, for example, if it received this as input:
10
2
1
20
The result of sorting would be:
1
10
2
20
Because that’s the alphabetical order of those characters. We can use the option sort -n to make sure it sorts these as numbers, in which case the output would be as expected:
1
2
10
20
Here’s the main message: always use the -n option if you want things that look like numbers to be sorted numerically (if the input doesn’t look like a number, then sort will just order them alphabetically instead).
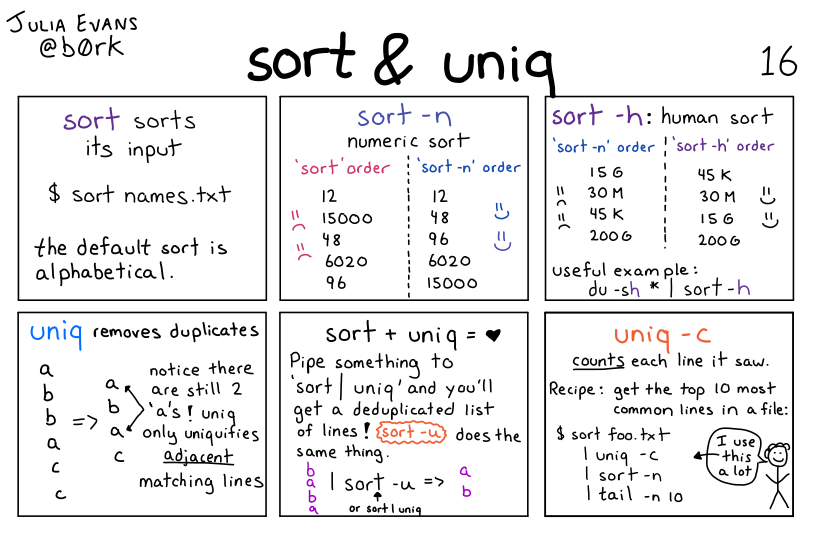
Illustration of the sort + uniq commands by Julia Evans
6.3 Exercises
Exercise 1 - Pipe comprehension
Level:
(Note: this is a conceptual exercise, you don’t need to use your own terminal.)
If you had the following two text files:
cat animals_1.txt
deer
rabbit
raccoon
rabbit
cat animals_2.txt
deer
fox
rabbit
bear
What would be the result of the following command?
cat animals*.txt |head-n 6 |tail-n 1
Answer
The result would be “fox”. Here is a diagram illustrating what happens in each of the three steps:
deer
rabbit deer
cat animals*.txt raccoon head -n 6 rabbit tail -n 1
-----------------> rabbit -----------> raccoon -----------> fox
deer rabbit
fox deer
rabbit fox
bear
cat animals*.txt would combine the content of both files, and then
head -n 6 would print the first six lines of the combined file, and then
tail -n 1 would return the last line of this output.
Exercise 2 - Sort & count
Level:
From the coronavirus/variants folder, let’s continue working on the command we looked at earlier to get the unique values in the variants column of our CSV files:
cat*_variants.csv |cut-d","-f 2 |sort|uniq
As you saw, this output also returns a line called “clade”. This was part of the header (column name) of our CSV file, which is not really useful to have in our output.
Let’s try and solve that problem, and also ask the question of how frequent each of these variants are in our data.
Looking at the help page for grep (man grep), see if you can find an option to invert a match, i.e. to return the lines that do not match a pattern. Can you think of how to include this in our pipeline to remove that line from our output?
Hint
The option to invert a match with grep is -v. Using one of our previous examples, grep -v "Alpha" would return the lines that do not match the word “Alpha”.
The uniq command has an option called -c. Try adding that option to the command and infer what it does (or look at man uniq).
Finally, produce a sorted table of counts for each of our variants in descending order (the most common variant at the top).
Hint
Look at the manual page for the sort command to find the option to order the output in reverse order (or do a quick web search). You may also want to use the -n option to sort numerically.
Answer
Task 1
Looking at the help of this function with man grep, we can find the following option:
-v, --invert-match select non-matching lines
So, we can continue working on our pipeline by adding another step at the end:
We used the option -r, which from the help page man sort, says:
-r, --reverse reverse the result of comparisons
We also used the -n option to ensure the result is sorted numerically. From the help page it says:
-n, --numeric-sort compare according to string numerical value
Exercise 3 - zcat and grep
Level:
In the coronavirus folder, you will find a file named proteins.fa.gz. This is a file that contains the amino acid sequences of the proteins in the SARS-CoV-2 coronavirus in a text-based format known as FASTA. However, this file is compressed using an algorithm known as GZip, which is indicated by the file extension .gz.
To look inside compressed files, you can use an alternative to cat called zcat (if you are using the macOS terminal use gzcat instead). The ‘z’ at the beggining indicates it will work on zipped files.
Use zcat (gzcat on macOS) together with less to look inside this file. Remember that you can press Q to exit the less program.
The content of this file may look a little strange, if you’re not familiar with the FASTA file format. Put simply, each protein sequence name starts with the > symbol. Combine zcat with grep to extract the sequence names only. How many proteins does SARS-CoV-2 have?
Answer
Task 1
The following command allows us to “browse” through the content of this file:
zcat proteins.fa.gz |less
We can use ↑ and ↓ to move line-by-line or the Page Up and Page Down keys to move page-by-page. You can exit less by pressing Q (for “quit”). This will bring you back to the console.
Task 2
We can look at the sequences’ names by running:
zcat proteins.fa.gz |grep">"
>ORF1ab protein=ORF1ab polyprotein
>ORF1ab protein=ORF1a polyprotein
>S protein=surface glycoprotein
>ORF3a protein=ORF3a protein
>E protein=envelope protein
>M protein=membrane glycoprotein
>ORF6 protein=ORF6 protein
>ORF7a protein=ORF7a protein
>ORF7b protein=ORF7b
>ORF8 protein=ORF8 protein
>N protein=nucleocapsid phosphoprotein
>ORF10 protein=ORF10 protein
We could further count how many sequences, by piping this output to wc:
zcat proteins.fa.gz |grep">"|wc-l
12
Exercise 4 - Counting values in columns
Level:
In the sequencing/ directory, you will find a file named gene_annotation.gtf.gz. This is a file containing the names and locations of genes in the Human genome in a standard text-based format called GTF.
This is a tab-delimited file, where the 3rd column contains information about the kind of annotated feature (e.g. a gene, an exon, start codon, etc.).
Using a combination of the commands we’ve seen so far:
Count how many occurrences of each feature (3rd column) there is in this file.
How many transcripts does the gene “ENSG00000113643” have?
Hint
Start by investigating the content of the file with zcat gene_annotation.gtf.gz | less -S. You will notice the first few lines of the file contain comments starting with # symbol. You should remove these lines before continuing.
Check the help for grep to remind yourself what the option is to return lines not matching a pattern.
Remember that the cut program uses tab as its default delimiter.
We use zcat because the file is compressed (we can tell from its extension ending with .gz).
We use grep to remove the first few lines of the file that start with # character.
We use cut to extract the third “field” (column) of the file. Because it’s a tab-delimited file, we don’t need to specify a delimiter with cut, as that is the default.
We use sort to order the features in alphabetical order.
Finally, uniq is used to return the unique values as well as count their occurrence (with the -c option).
Task 2
The answer is 10. We could use the following command: