# Load required R libraries
library(tidyverse)
library(performance)
# Read in the required data
islands <- read_csv("data/islands.csv")
seatbelts <- read_csv("data/seatbelts.csv")12 Analysing count data
Learning outcomes
- Be able to identify count data
- Fit a Poisson regression model with count data
- Assess whether assumptions are met and test significance of the model
12.1 Context
We have now covered analysing binary responses and learned how to assess the quality and appropriateness of the resulting models. In the next sections we extend this further, by looking at a different type of response variable: count data. The humble count may look innocent, but often hides a whole lot of nuances that are hard to spot. So, pay attention!
12.2 Section setup
Click to expand
We’ll use the following libraries and data:
# Load required Python libraries
import pandas as pd
import math
import statsmodels.api as sm
import statsmodels.formula.api as smf
from scipy.stats import *
from plotnine import *
# Read in the required data
islands = pd.read_csv("data/islands.csv")
seatbelts = pd.read_csv("data/seatbelts.csv")The worked example in this chapter will use the islands dataset.
This is a dataset comprising 35 observations of two variables. For each small island in the dataset, the researcher recorded the number of unique species; they want to know if this can be predicted from the area (km2) of the island.
12.3 Load and visualise the data
First we load the data, then we visualise it.
islands <- read_csv("data/islands.csv")
head(islands)# A tibble: 6 × 2
species area
<dbl> <dbl>
1 114 12.1
2 130 13.4
3 113 13.7
4 109 14.5
5 118 16.8
6 136 19.0islands = pd.read_csv("data/islands.csv")
islands.head() species area
0 114 12.076133
1 130 13.405439
2 113 13.723525
3 109 14.540359
4 118 16.792122We can plot the data:
ggplot(islands, aes(x = area, y = species)) +
geom_point()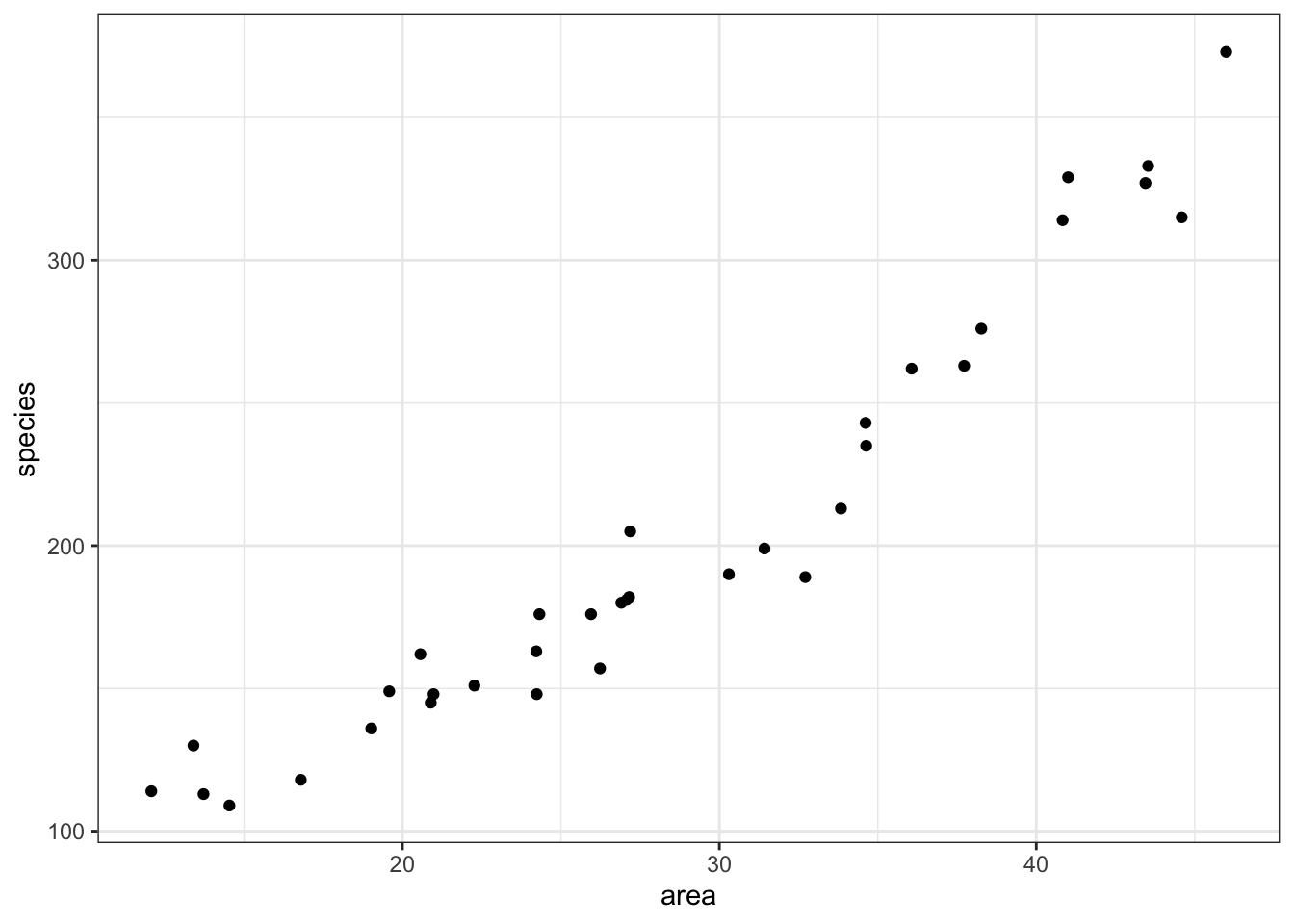
p = (ggplot(islands, aes(x = "area", y = "species")) +
geom_point())
p.show()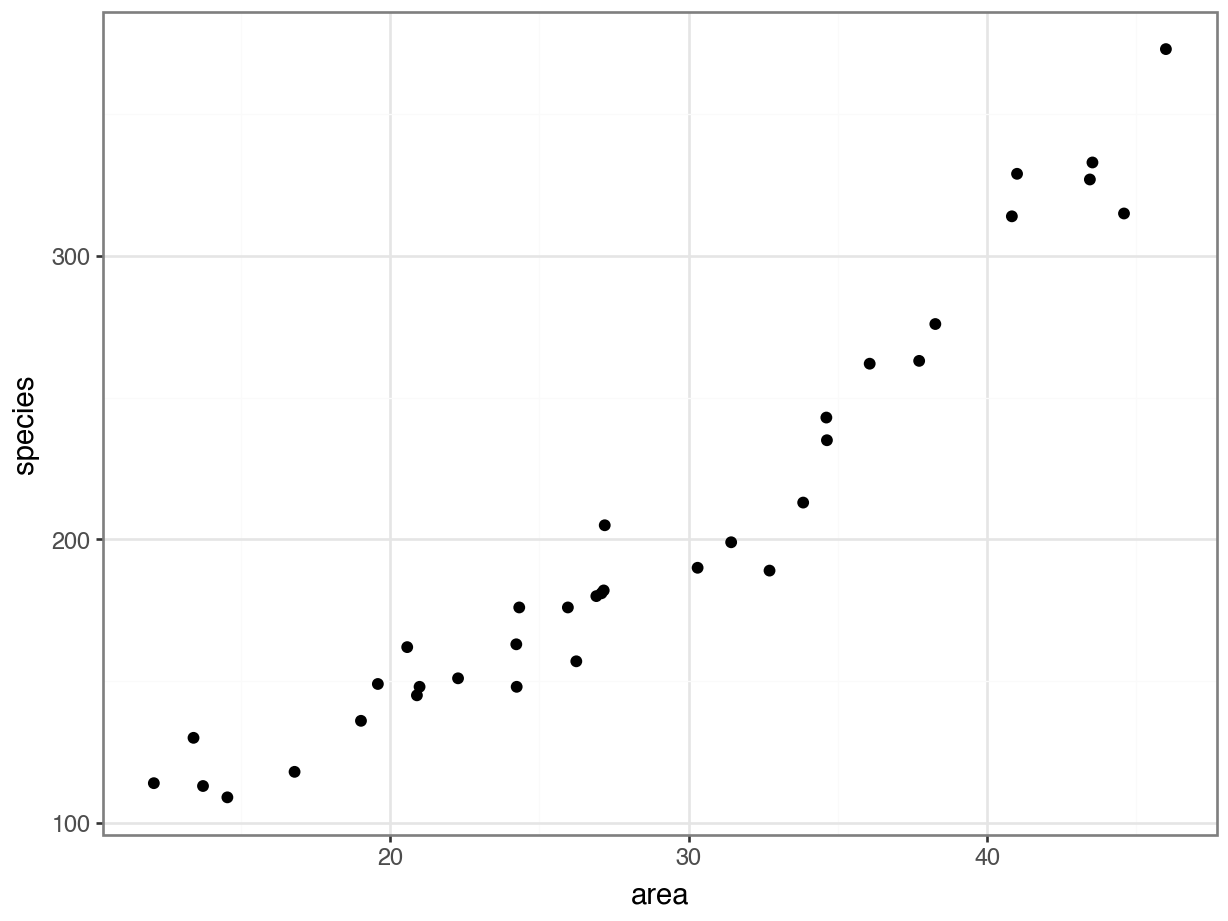
Each dot on the scatterplot represents an island in the dataset.
It looks as though area definitely has some relationship with the number of species that we observe.
Next step is to try to model these data.
12.4 Constructing a model
The species variable is the outcome or response (since we’re interested in whether it’s predicted by area).
It qualifies as a count variable. It’s bounded at 0 and \(\infty\), and can only jump up in integers - in other words, you can’t have less than 0 species, or 6.3 species.
This means the best place to start is a Poisson regression. We fit these in a very similar way to a logistic regression, just with a different option specified for the family argument.
glm_isl <- glm(species ~ area,
data = islands, family = "poisson")and we look at the model summary:
summary(glm_isl)
Call:
glm(formula = species ~ area, family = "poisson", data = islands)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 4.241129 0.041322 102.64 <2e-16 ***
area 0.035613 0.001247 28.55 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for poisson family taken to be 1)
Null deviance: 856.899 on 34 degrees of freedom
Residual deviance: 30.437 on 33 degrees of freedom
AIC: 282.66
Number of Fisher Scoring iterations: 3The output is strikingly similar to the logistic regression models (who’d have guessed, eh?) and the main numbers to extract from the output are the coefficients (the two numbers underneath Estimate in the table above):
coefficients(glm_isl)(Intercept) area
4.24112904 0.03561346 model = smf.glm(formula = "species ~ area",
family = sm.families.Poisson(),
data = islands)
glm_isl = model.fit()Let’s look at the model output:
print(glm_isl.summary()) Generalized Linear Model Regression Results
==============================================================================
Dep. Variable: species No. Observations: 35
Model: GLM Df Residuals: 33
Model Family: Poisson Df Model: 1
Link Function: Log Scale: 1.0000
Method: IRLS Log-Likelihood: -139.33
Date: Fri, 25 Jul 2025 Deviance: 30.437
Time: 08:28:46 Pearson chi2: 30.3
No. Iterations: 4 Pseudo R-squ. (CS): 1.000
Covariance Type: nonrobust
==============================================================================
coef std err z P>|z| [0.025 0.975]
------------------------------------------------------------------------------
Intercept 4.2411 0.041 102.636 0.000 4.160 4.322
area 0.0356 0.001 28.551 0.000 0.033 0.038
==============================================================================The output is strikingly similar to the logistic regression models (who’d have guessed, eh?) and the main numbers to extract from the output are the coefficients (the two numbers underneath coef in the table above):
print(glm_isl.params)Intercept 4.241129
area 0.035613
dtype: float6412.4.1 The model equation
Now that we have the beta coefficients, we can place them inside an equation.
The left-hand side of this equation is the expected number of species, \(E(species)\).
On the right-hand side, we take the linear equation \(\beta_0 + \beta_1 * x_1\) and embed it inside the inverse link function, which in this case is just the exponential function.
It looks like this:
\[ E(species) = \exp(4.24 + 0.036 \times area) \]
Interpreting this requires a bit of thought (not much, but a bit).
The intercept coefficient, 4.24, is related to the number of species we would expect on an island of zero area. But in order to turn this number into something meaningful we have to exponentiate it.
Since \(\exp(4.24) \approx 69\), we can say that the baseline number of species the model expects on any island is 69.
The coefficient of area is the fun bit. For starters we can see that it is a positive number which does mean that increasing area leads to increasing numbers of species. Good so far.
But what does the value 0.036 actually mean? Well, if we exponentiate it as well, we get \(\exp(0.036) \approx 1.04\). This means that for every increase in area of 1 km2 (the original units of the area variable), the number of species on the island is multiplied by 1.04.
So, an island of area km2 will have \(1.04 \times 69 \approx 72\) species.
12.5 Plotting the Poisson regression
ggplot(islands, aes(area, species)) +
geom_point() +
geom_smooth(method = "glm", se = FALSE, fullrange = TRUE,
method.args = list(family = "poisson")) +
xlim(10,50)`geom_smooth()` using formula = 'y ~ x'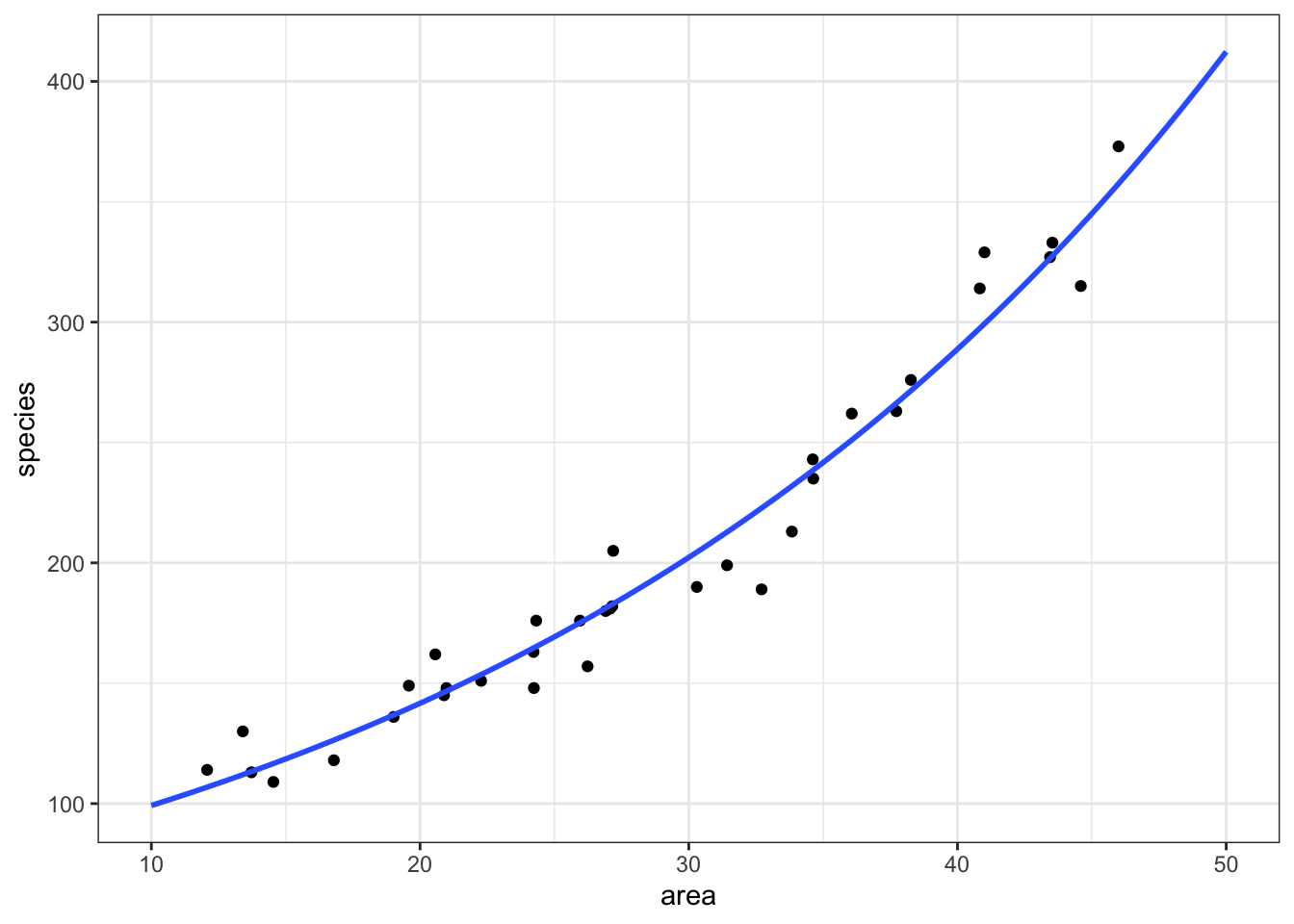
species ~ area
First, we produce a set of predictions, which we can then plot.
model = pd.DataFrame({'area': list(range(10, 50))})
model["pred"] = glm_isl.predict(model)
model.head() area pred
0 10 99.212463
1 11 102.809432
2 12 106.536811
3 13 110.399326
4 14 114.401877p = (ggplot(islands, aes(x = "area", y = "species")) +
geom_point() +
geom_line(model, aes(x = "area", y = "pred"),
colour = "blue", size = 1))
p.show()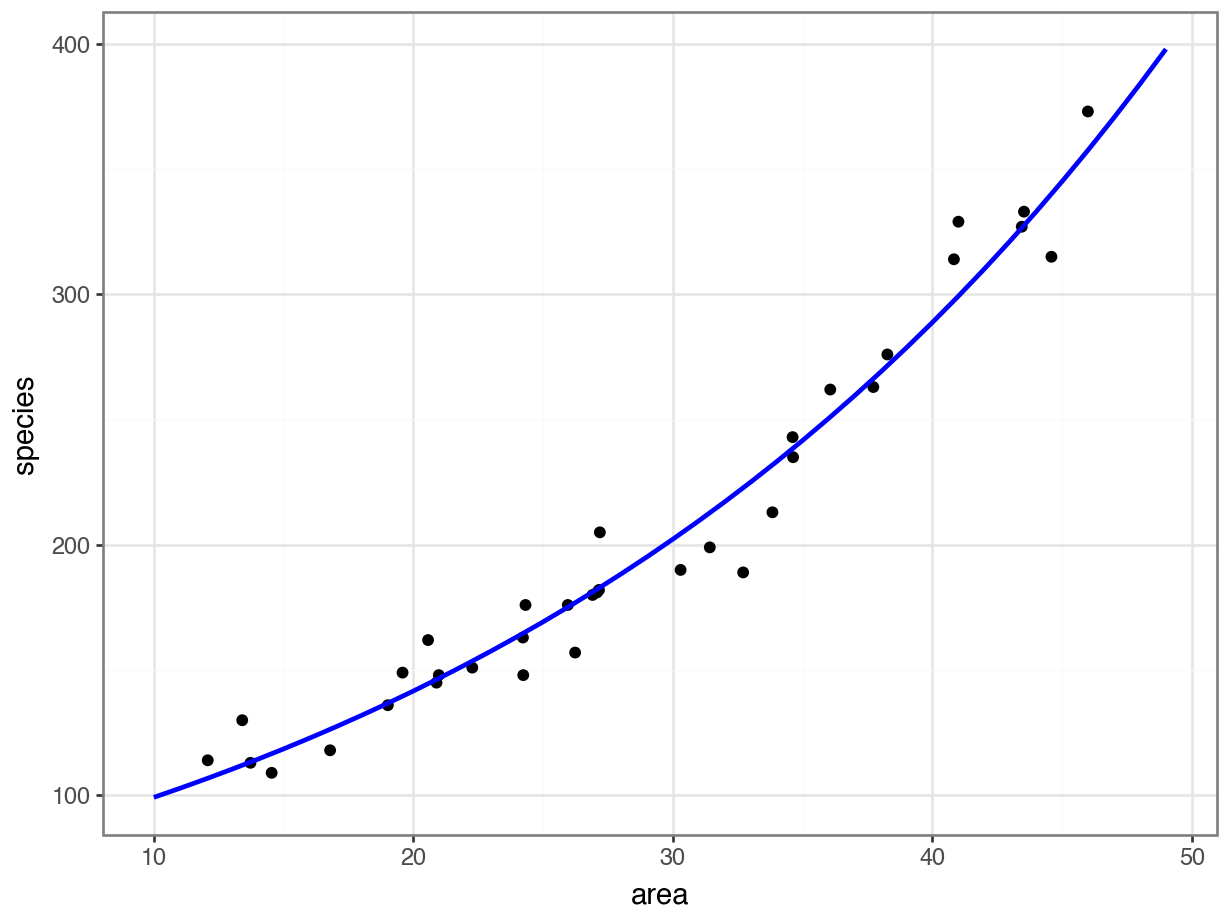
species ~ area
12.6 Assumptions & model fit
As a reminder from last chapter, we want to consider the following things to assess whether we’ve fit the right model:
- The distribution of the response variable
- The link function
- Independence
- Influential points
(We don’t need to worry about collinearity here - there’s only one predictor!)
We’re also going to talk about dispersion, which really comes into play when talking about count data - we’ll get to that after we’ve run through the stuff that’s familiar.
With the generic check_model function, we can visually assess a few things quite quickly.
check_model(glm_isl)Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.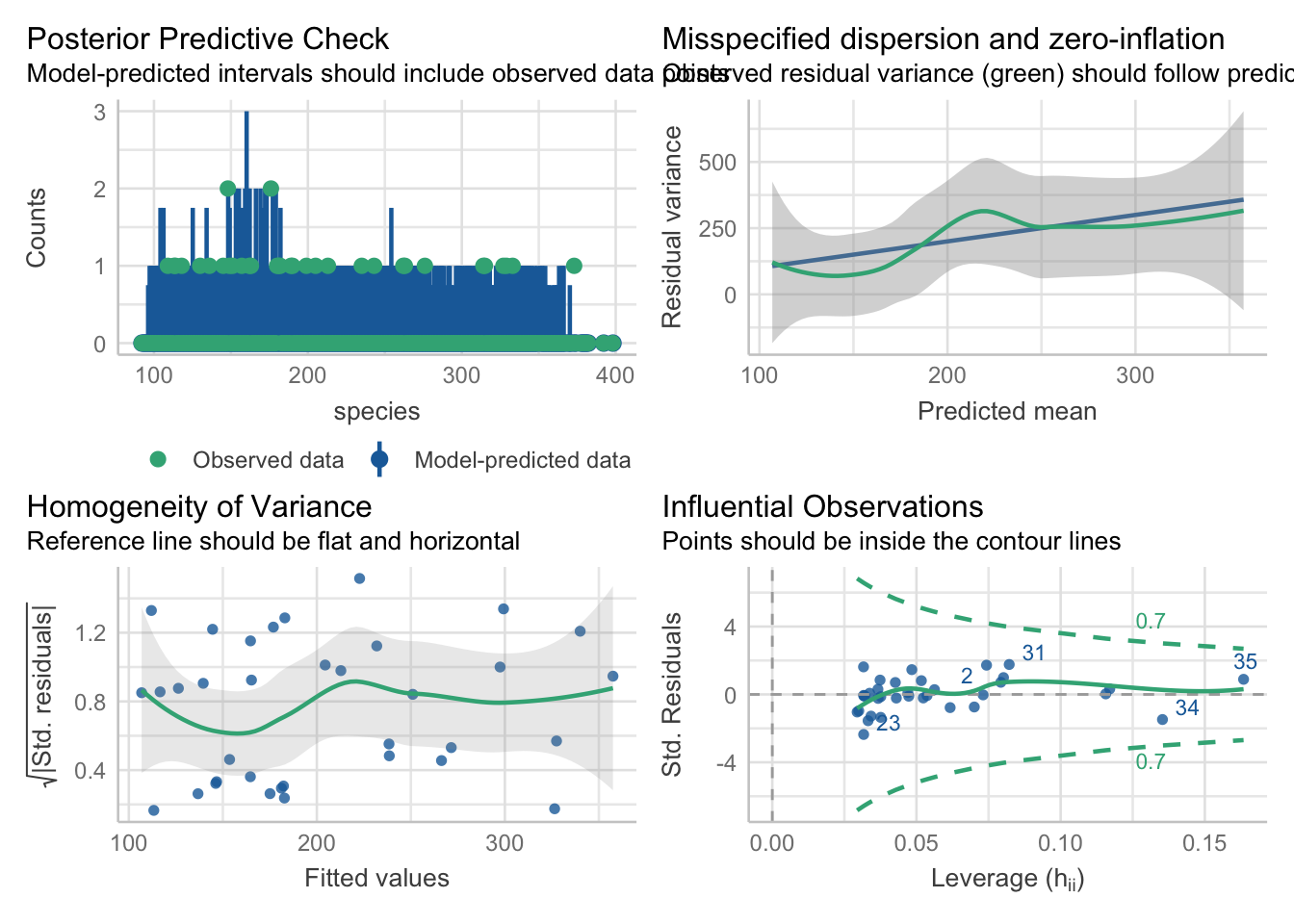
glm_isl
The posterior predictive check looks alright. The blue simulations seem to be following a pretty similar distribution to the actual data in green.
The leverage/Cook’s distance plot also isn’t identifying any data points that we need to be concerned about. We can follow up on that:
check_outliers(glm_isl, threshold = list('cook'=0.5))OK: No outliers detected.
- Based on the following method and threshold: cook (0.5).
- For variable: (Whole model)We can check for influential points:
influence = glm_isl.get_influence()
cooks = influence.cooks_distance[0]
influential_points = np.where(cooks > 0.5)[0]
print("Influential points:", influential_points)Influential points: []Influential points: There’s no evidence of any points with high Cook’s distances, so life is rosy on that front.
Independence: From the description of the dataset, it sounds plausible that each island is independent. Of course, if we later found out that some of the islands were clustered together into a bunch of archipelagos, that would definitely be cause for concern.
Distribution & link function: Again, from the description of the species variable, we can be confident that this really is a count variable. Whether or not Poisson regression with the log link function is correct, however, will depend on what happens when we look at the dispersion parameter.
12.7 Dispersion
12.7.1 What is dispersion?
Dispersion, in statistics, is a general term to describe the variability, scatter, or spread of a distribution. Variance is actually a type of dispersion.
In a normal distribution, the mean (average/central tendency) and the variance (dispersion) are independent of each other; we need both numbers, or parameters, to understand the shape of the distribution.
Other distributions, however, require different parameters to describe them in full. For a Poisson distribution - which we’ll learn more about when we talk about count data - we need just one parameter (\(\lambda\)) to describe the distribution, because the mean and variance are assumed to be the same.
In the context of a model, you can think about the dispersion as the degree to which the data are spread out around the model curve. A dispersion parameter of 1 means the data are spread out exactly as we expect; <1 is called underdispersion; and >1 is called overdispersion.
12.7.3 Assessing dispersion
The easiest way to check dispersion in a model is to calculate the ratio of the residual deviance to the residual degrees of freedom.
If we take a look at the model output, we can see the two quantities we care about - residual deviance and residual degrees of freedom:
summary(glm_isl)
Call:
glm(formula = species ~ area, family = "poisson", data = islands)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 4.241129 0.041322 102.64 <2e-16 ***
area 0.035613 0.001247 28.55 <2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for poisson family taken to be 1)
Null deviance: 856.899 on 34 degrees of freedom
Residual deviance: 30.437 on 33 degrees of freedom
AIC: 282.66
Number of Fisher Scoring iterations: 3print(glm_isl.summary()) Generalized Linear Model Regression Results
==============================================================================
Dep. Variable: species No. Observations: 35
Model: GLM Df Residuals: 33
Model Family: Poisson Df Model: 1
Link Function: Log Scale: 1.0000
Method: IRLS Log-Likelihood: -139.33
Date: Fri, 25 Jul 2025 Deviance: 30.437
Time: 08:28:47 Pearson chi2: 30.3
No. Iterations: 4 Pseudo R-squ. (CS): 1.000
Covariance Type: nonrobust
==============================================================================
coef std err z P>|z| [0.025 0.975]
------------------------------------------------------------------------------
Intercept 4.2411 0.041 102.636 0.000 4.160 4.322
area 0.0356 0.001 28.551 0.000 0.033 0.038
==============================================================================The residual deviance is 30.44, on 33 residual degrees of freedom. All we need to do is divide one by the other to get our dispersion parameter.
glm_isl$deviance/glm_isl$df.residual[1] 0.922334print(glm_isl.deviance/glm_isl.df_resid)0.9223340414458482The dispersion parameter here is 0.9223. That’s pretty good - not far off 1 at all.
But how can we check whether it is significantly different from 1?
Once again, the performance package comes in very helpful here.
The check_overdispersion function will give us both the dispersion parameter and a p-value (which is based on a chi-square test - like the goodness-of-fit ones you were running last chapter).
check_overdispersion(glm_isl)# Overdispersion test
dispersion ratio = 0.919
Pearson's Chi-Squared = 30.339
p-value = 0.6No overdispersion detected.Here, it confirms our suspicions; the dispersion parameter is both descriptively close to 1, and also not significantly different from 1.
You’ll notice that this function does give a slightly different value for the dispersion parameter, compared to when we calculated it manually above.
This is because it actually uses the sum of squared Pearson residuals instead of the residual deviance (a distinction which really isn’t worth worrying about). Broadly, the conclusion should be the same, so the difference doesn’t matter very much.
Well, you’ve actually already got the knowledge you need to do this, from the previous chapter on significance testing.
Specifically, the chi-squared goodness-of-fit test can be used to check whether the dispersion is within sensible limits.
pvalue = chi2.sf(glm_isl.deviance, glm_isl.df_resid)
print(pvalue)0.5953470127463268If our chi-squared goodness-of-fit test returns a large (insignificant) p-value, as it does here, that tells us that we don’t need to worry about the dispersion.
If our chi-squared goodness-of-fit test returned a small, significant p-value, this would tell us our model doesn’t fit the data well. And, since dispersion is all about the spread of points around the model, it makes sense that these two things are so closely related!
Great - we can proceed with the Poisson regression, since we don’t seem to have overdispersed or underdispersed data.
Next chapter we’ll look at what we would have done next, if we had run into problems with dispersion.
12.8 Assessing significance
By testing the dispersion with a chi-square test, we have already essentially checked the goodness-of-fit of this model - it’s good.
This leaves us with two things to check:
- Is the overall model better than the null model?
- Are any of the individual predictors significant?
12.8.1 Comparing against the null
We need to fit the null model, and then we can extract the null deviance and degrees of freedom to compare against our model.
glm_isl_null <- glm(species ~ 1,
data = islands, family = "poisson")
anova(glm_isl, glm_isl_null)Analysis of Deviance Table
Model 1: species ~ area
Model 2: species ~ 1
Resid. Df Resid. Dev Df Deviance Pr(>Chi)
1 33 30.44
2 34 856.90 -1 -826.46 < 2.2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Since there’s only one predictor, we actually could achieve the same effect here by just computing an analysis of deviance table:
anova(glm_isl, test = "Chisq")Analysis of Deviance Table
Model: poisson, link: log
Response: species
Terms added sequentially (first to last)
Df Deviance Resid. Df Resid. Dev Pr(>Chi)
NULL 34 856.90
area 1 826.46 33 30.44 < 2.2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1First, we fit a null model:
model = smf.glm(formula = "species ~ 1",
family = sm.families.Poisson(),
data = islands)
glm_isl_null = model.fit()
glm_isl_null.df_residnp.int64(34)And now we can compare the two:
# Calculate the likelihood ratio (i.e. the chi-square value)
lrstat = -2*(glm_isl_null.llf - glm_isl.llf)
# Calculate the associated p-value
pvalue = chi2.sf(lrstat, glm_isl_null.df_resid - glm_isl.df_resid)
print(lrstat, pvalue)826.4623291533155 9.523357725017566e-182This gives a reported p-value extremely close to zero, which is rather small.
Therefore, this model is significant over the null, and species does appear to be predicted by area.
12.9 Exercises
12.9.1 Seat belts
Exercise 1 - Seat belts
Level:
For this exercise we’ll be using the data from data/seatbelts.csv.
The data tracks the number of drivers killed in road traffic accidents, before and after the seat belt law was introduced. The information on whether the law was in place is encoded in the law column as 0 (law not in place) or 1 (law in place).
The year variable is our predictor of interest.
In this exercise, you should:
- Visualise the data
- Create a poisson regression model and extract its equation
- Plot the regression model on top of the data
- Assess if the model is a decent predictor for the number of fatalities (& check assumptions)
seatbelts <- read_csv("data/seatbelts.csv")
head(seatbelts)# A tibble: 6 × 10
casualties drivers front rear kms petrol_price van_killed law year month
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
1 107 1687 867 269 9059 0.103 12 0 1969 Jan
2 97 1508 825 265 7685 0.102 6 0 1969 Feb
3 102 1507 806 319 9963 0.102 12 0 1969 Mar
4 87 1385 814 407 10955 0.101 8 0 1969 Apr
5 119 1632 991 454 11823 0.101 10 0 1969 May
6 106 1511 945 427 12391 0.101 13 0 1969 Jun seatbelts = pd.read_csv("data/seatbelts.csv")
seatbelts.head() casualties drivers front rear ... van_killed law year month
0 107 1687 867 269 ... 12 0 1969 Jan
1 97 1508 825 265 ... 6 0 1969 Feb
2 102 1507 806 319 ... 12 0 1969 Mar
3 87 1385 814 407 ... 8 0 1969 Apr
4 119 1632 991 454 ... 10 0 1969 May
[5 rows x 10 columns]
Answer
Visualise the data
First we have a look at the data comparing no law versus law:
We have to convert the law column to a factor, otherwise R will see it as numerical.
seatbelts |>
ggplot(aes(as_factor(law), casualties)) +
geom_boxplot() +
geom_jitter(width = 0.2)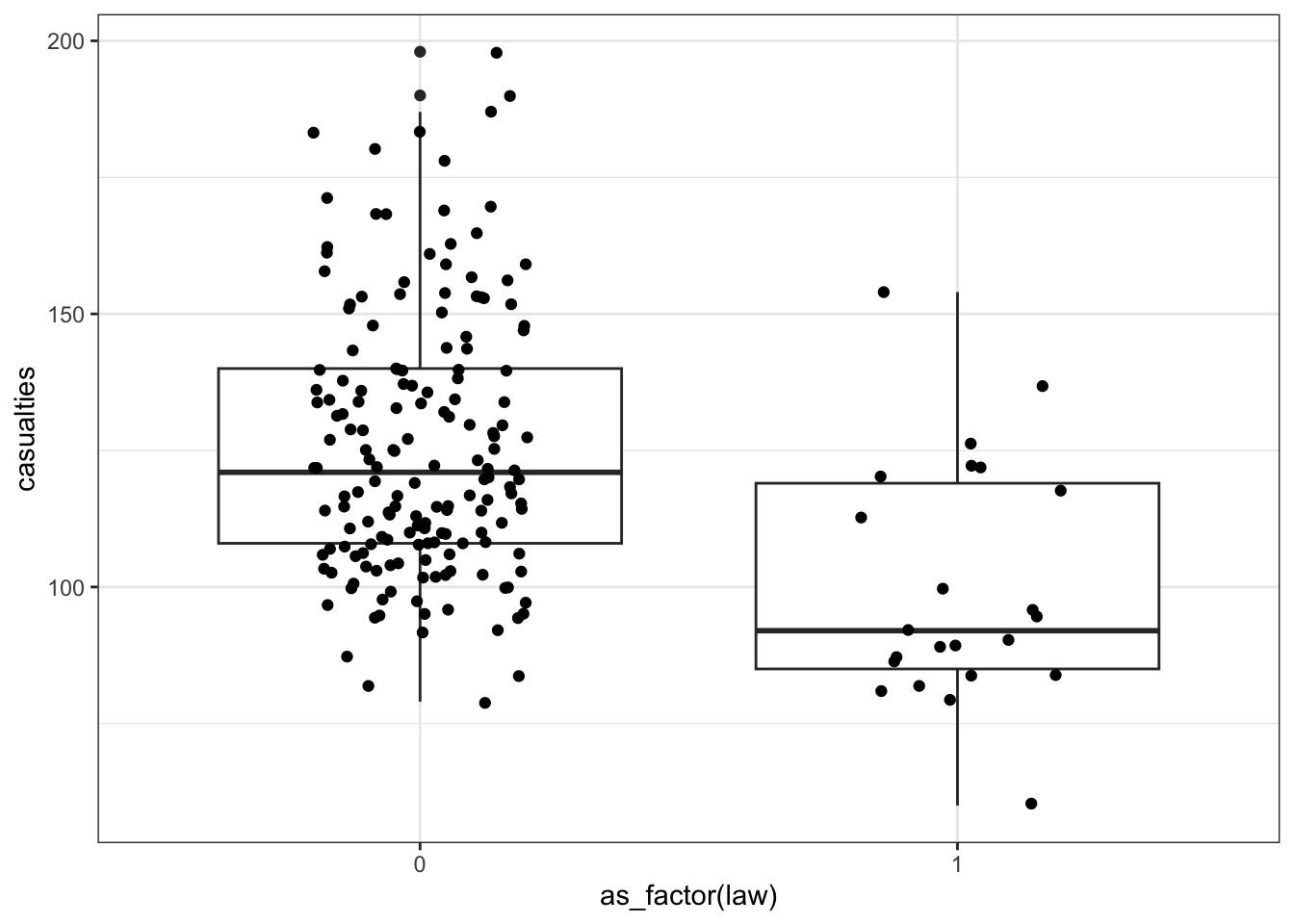
The data are recorded by month and year, so we can also display the number of drivers killed by year:
seatbelts |>
ggplot(aes(year, casualties)) +
geom_point()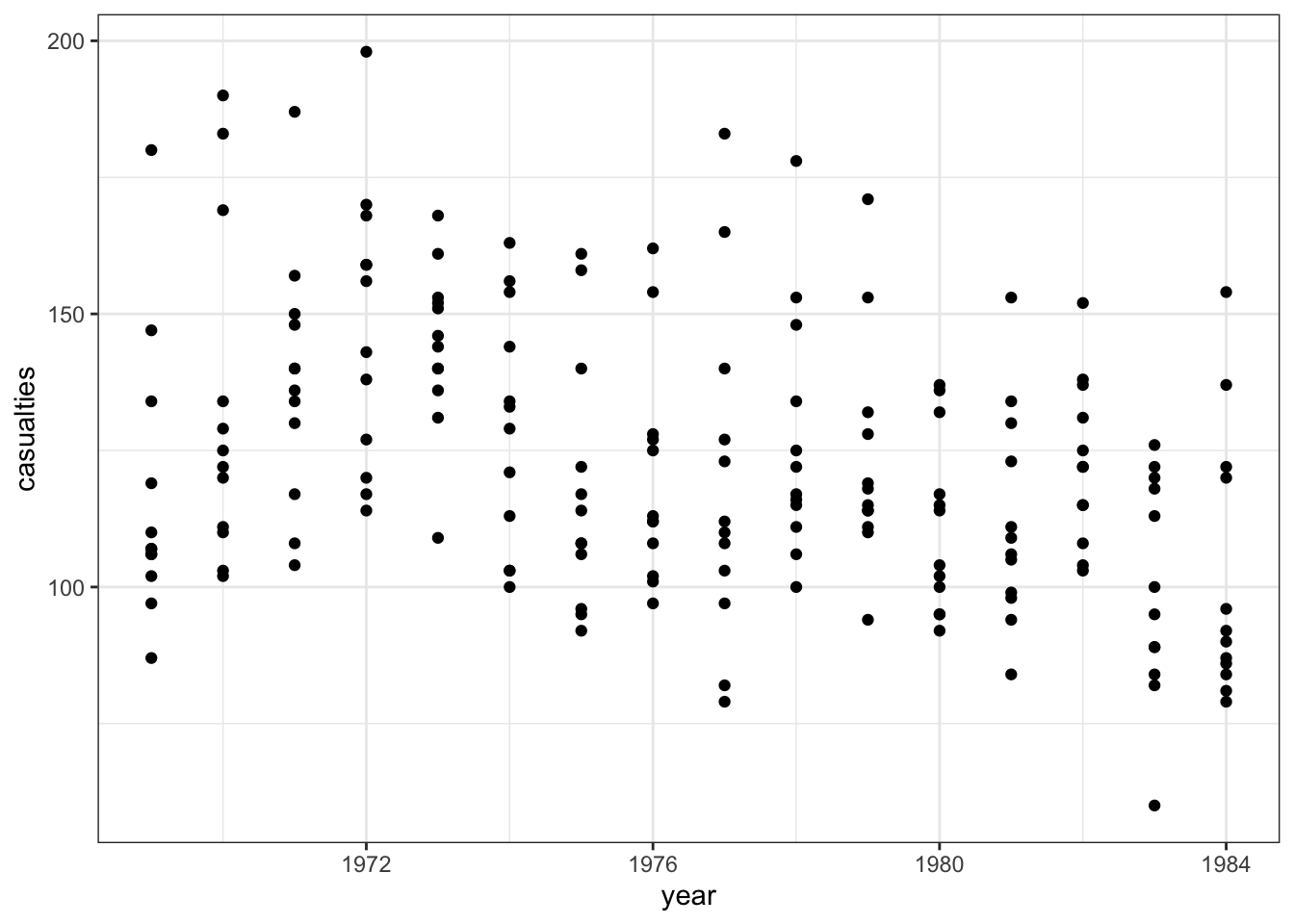
We have to convert the law column to a factor, otherwise R will see it as numerical.
p = (ggplot(seatbelts, aes(x = seatbelts.law.astype(object),
y = "casualties")) +
geom_boxplot() +
geom_jitter(width = 0.2))
p.show()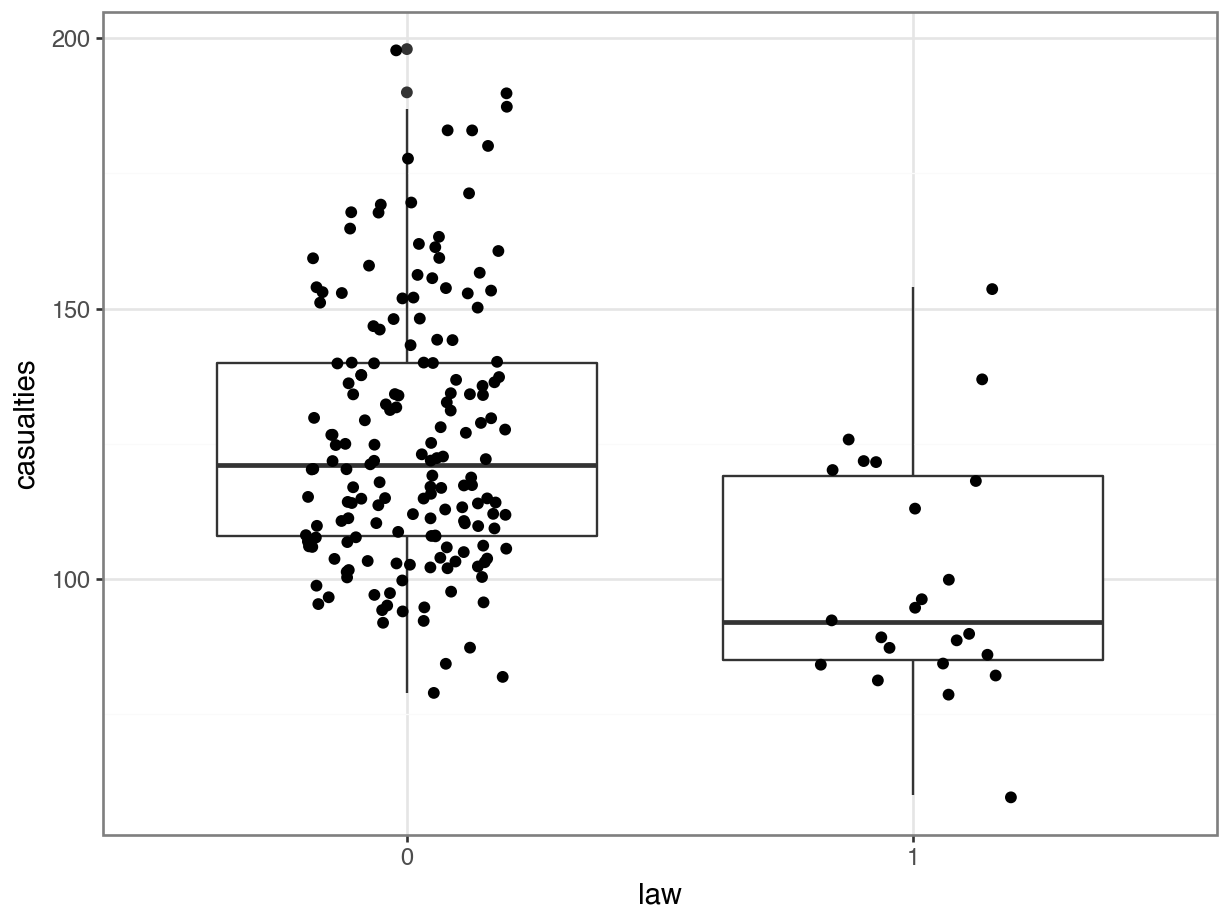
The data are recorded by month and year, so we can also display the number of casualties by year:
p = (ggplot(seatbelts,
aes(x = "year",
y = "casualties")) +
geom_point())
p.show()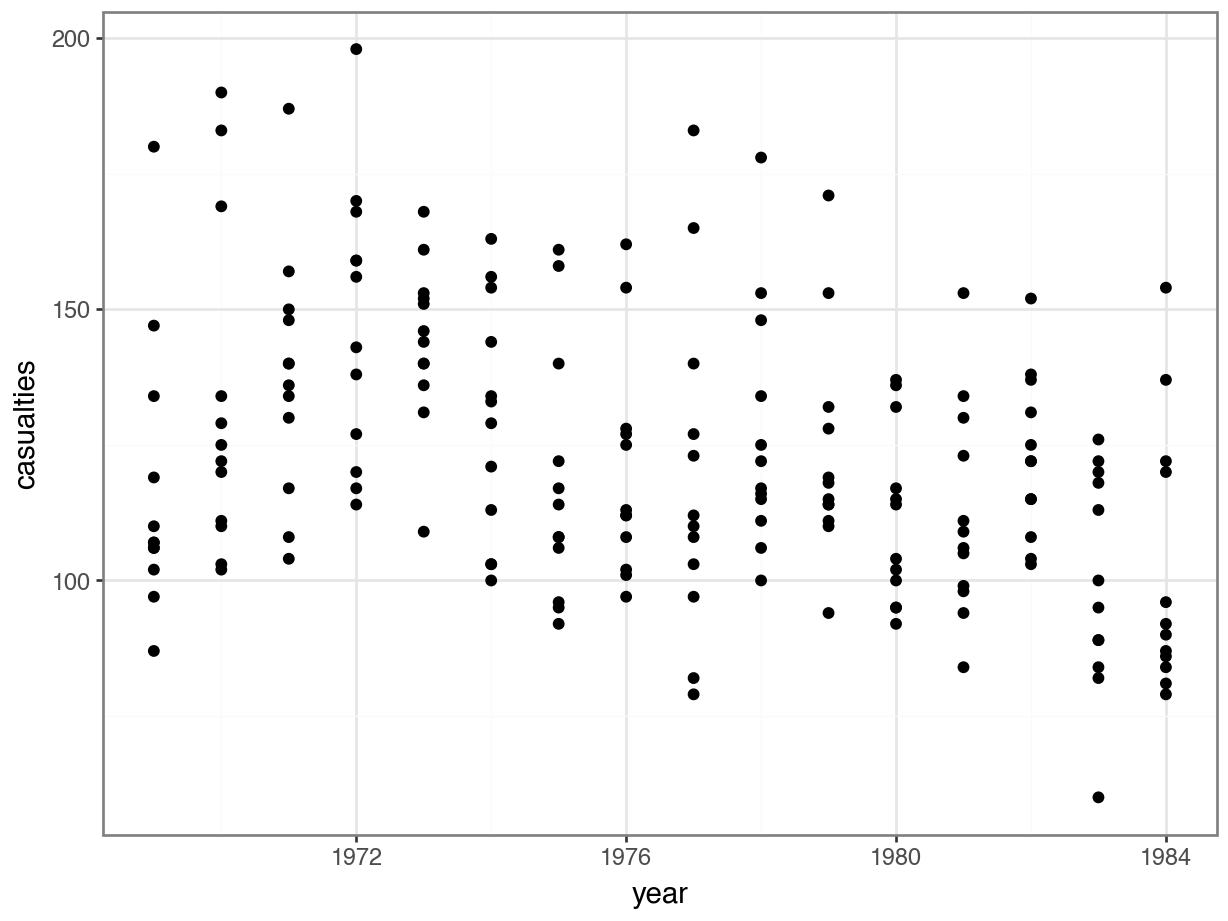
The data look a bit weird. There’s a bit of a wavy pattern across years. And although it looks like fatalities are lower after the law was implemented, there are many more observations when the law was not in place, which is going to make the data harder to interpret.
Constructing a model
glm_stb <- glm(casualties ~ year,
data = seatbelts, family = "poisson")model = smf.glm(formula = "casualties ~ year",
family = sm.families.Poisson(),
data = seatbelts)
glm_stb = model.fit()Model equation
coefficients(glm_stb)(Intercept) year
37.168958 -0.016373 print(glm_stb.params)Intercept 37.168958
year -0.016373
dtype: float64The coefficients of the Poisson model equation should be placed in the following formula, in order to estimate the expected number of species as a function of island size:
\[ E(casualties) = \exp(37.17 - -0.016 \times year) \]
Visualise model
We can just adapt the code used in the worked example earlier in the chapter:
ggplot(seatbelts, aes(year, casualties)) +
geom_point() +
geom_smooth(method = "glm", se = FALSE, fullrange = TRUE,
method.args = list(family = poisson)) +
xlim(1970, 1985)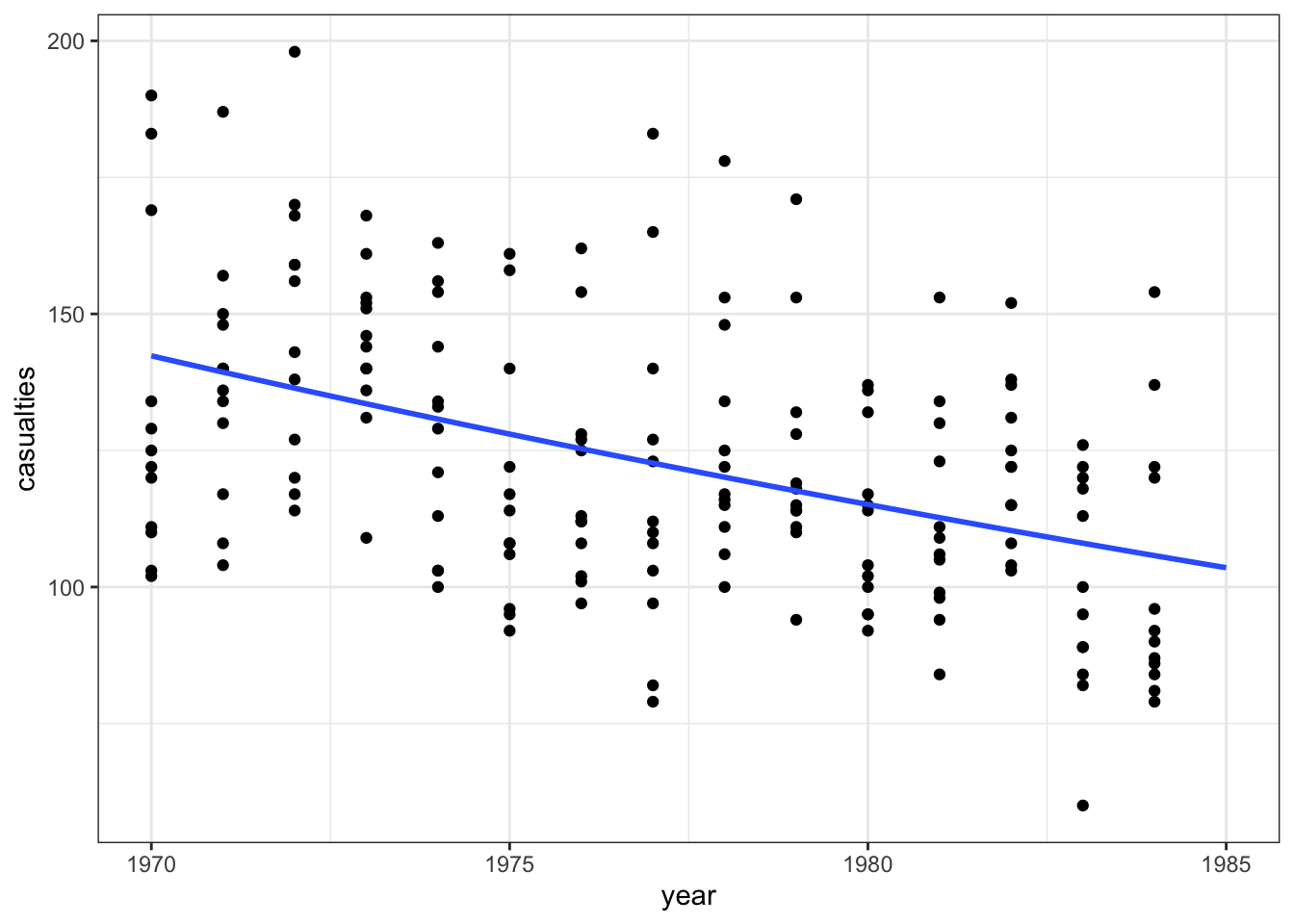
model = pd.DataFrame({'year': list(range(1968, 1985))})
model["pred"] = glm_stb.predict(model)
model.head() year pred
0 1968 140.737690
1 1969 138.452153
2 1970 136.203733
3 1971 133.991827
4 1972 131.815842p = (ggplot(seatbelts, aes(x = "year",
y = "casualties")) +
geom_point() +
geom_line(model, aes(x = "year", y = "pred"),
colour = "blue", size = 1))
p.show()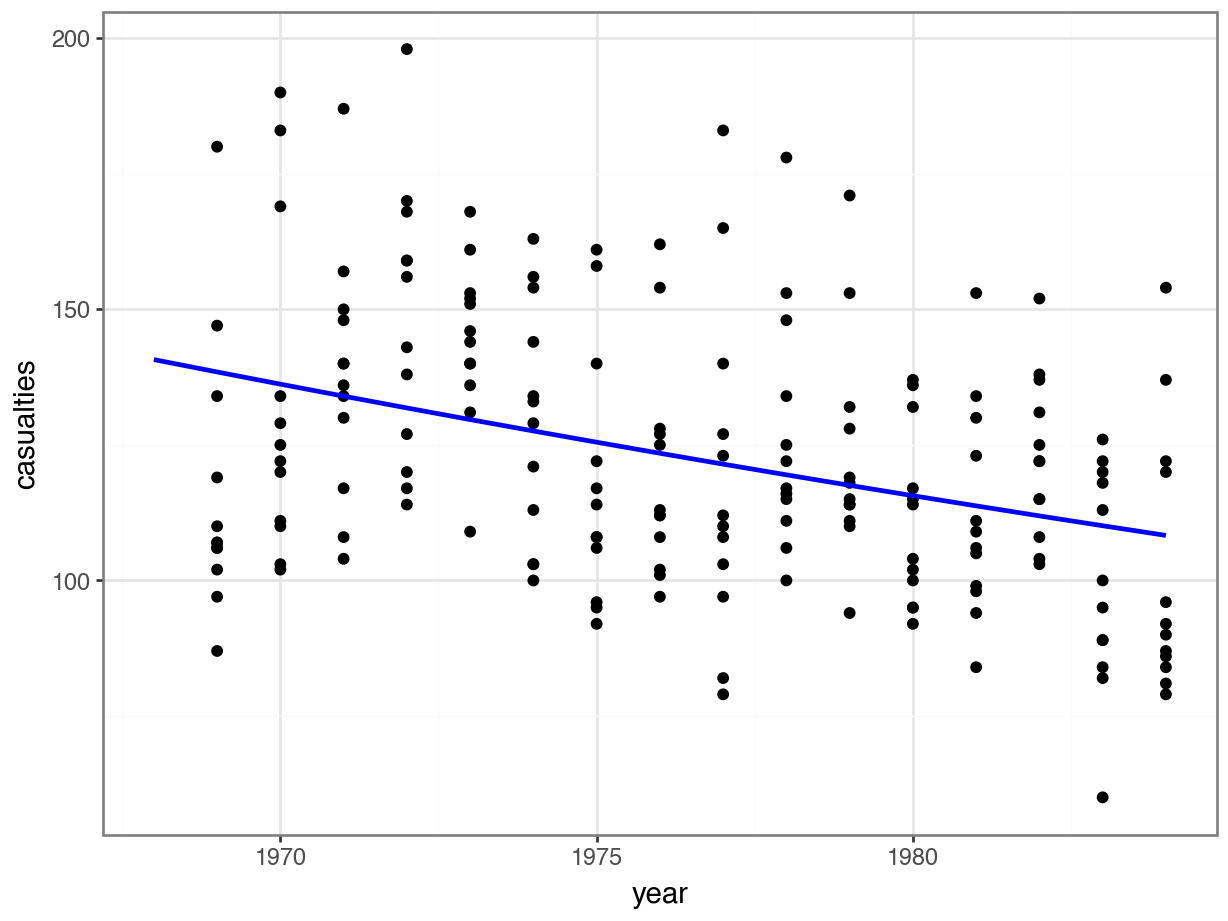
Assessing model quality & significance
Are the assumptions met?
| Response distribution & link function | Yes, based on the info given |
| Independence | Yes, based on the info given |
| Influential points | Check with Cook’s distance |
| Dispersion | Check dispersion parameter |
We can assess outliers visually or by printing a list of high leverage points:
check_outliers(glm_stb, threshold = list('cook'=0.5))OK: No outliers detected.
- Based on the following method and threshold: cook (0.5).
- For variable: (Whole model)check_model(glm_stb, check = 'outliers')Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.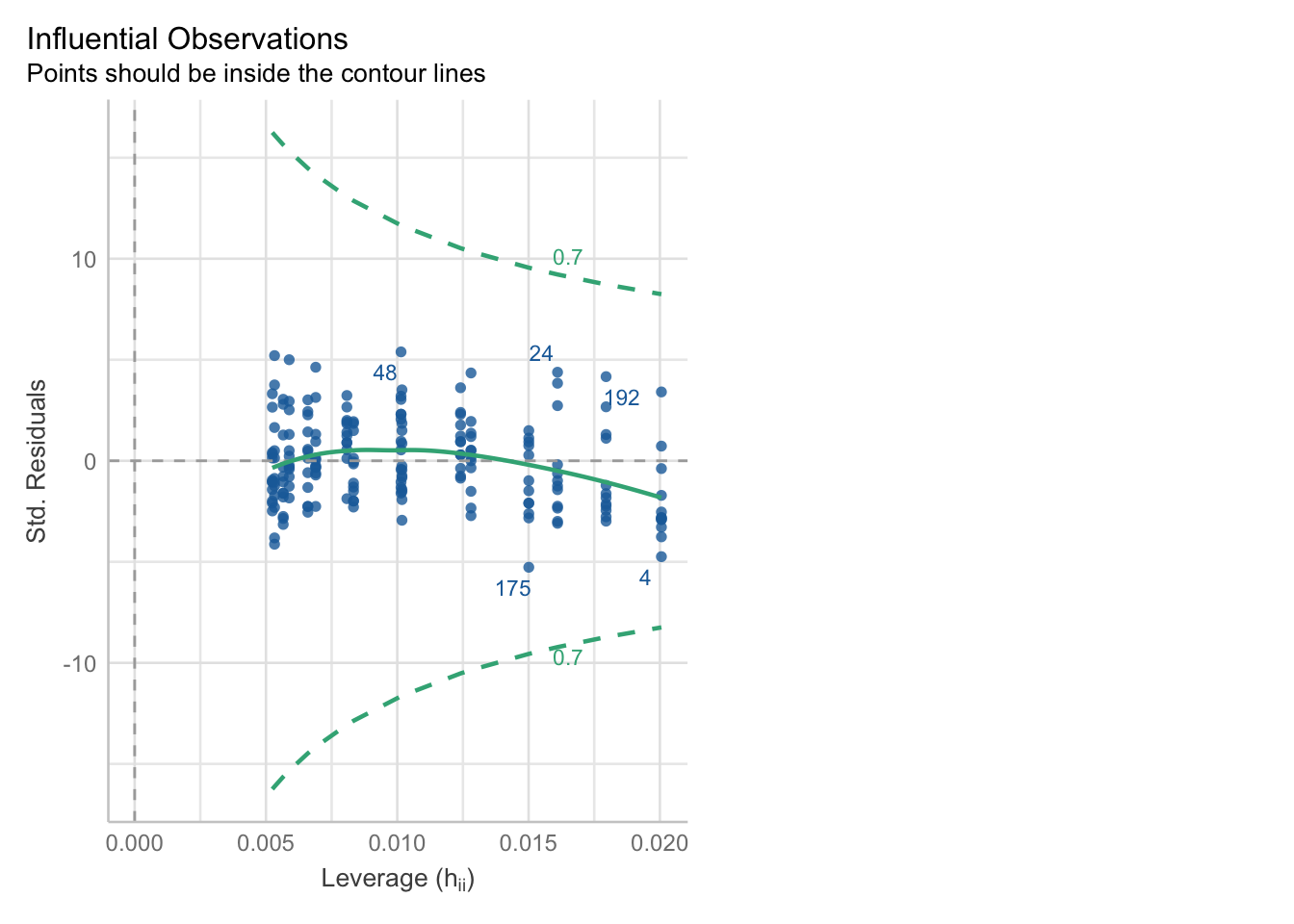
It seems we’re okay on the outlier front.
For dispersion, again we have some options of how to test it:
check_overdispersion(glm_stb)# Overdispersion test
dispersion ratio = 4.535
Pearson's Chi-Squared = 861.667
p-value = < 0.001Overdispersion detected.check_model(glm_stb, check = 'overdispersion')Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.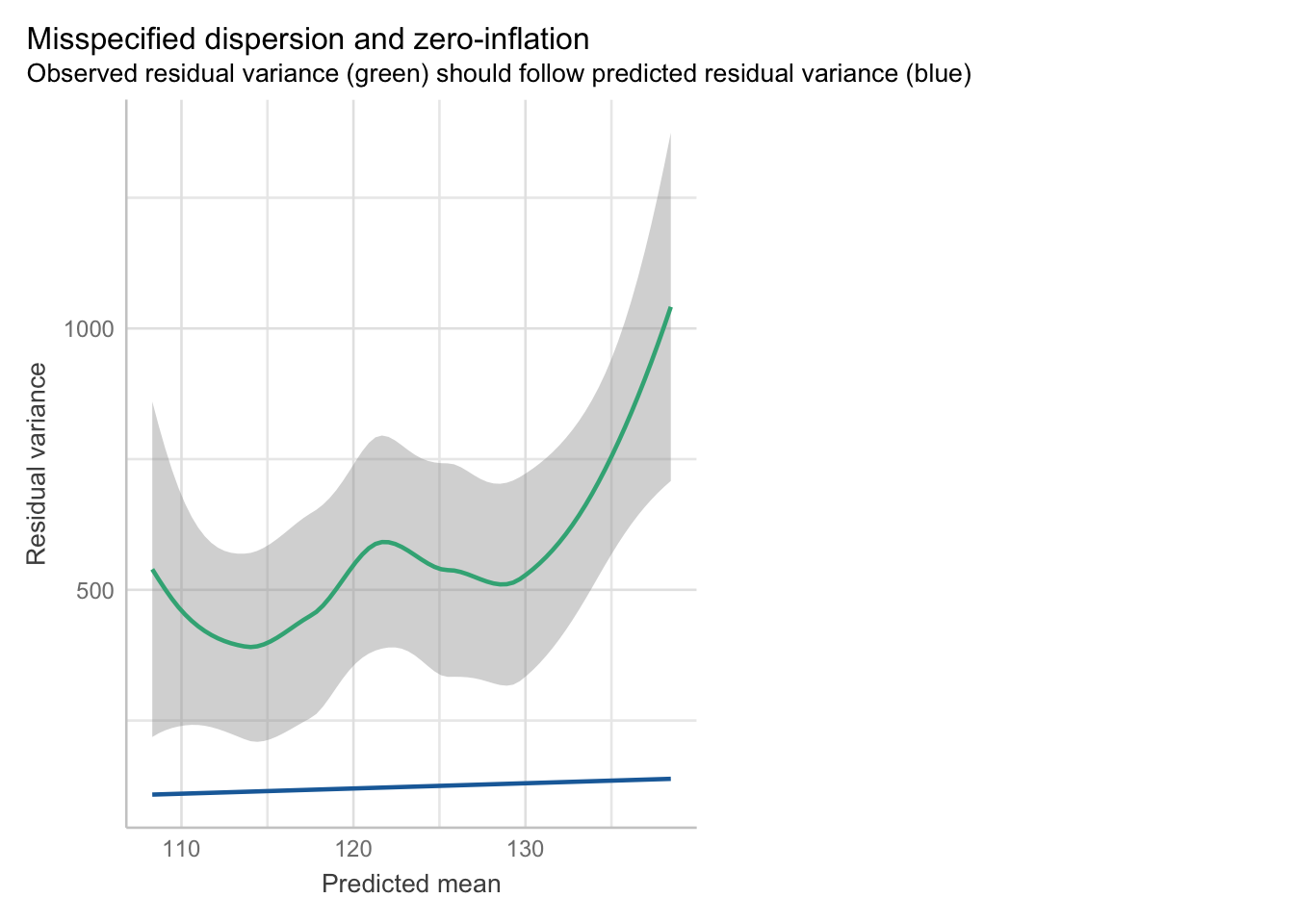
Dispersion is definitely a problem here.
For completeness, we can also peek at the posterior predictive check:
check_model(glm_stb, check = 'pp_check')Cannot simulate residuals for models of class `glm`. Please try
`check_model(..., residual_type = "normal")` instead.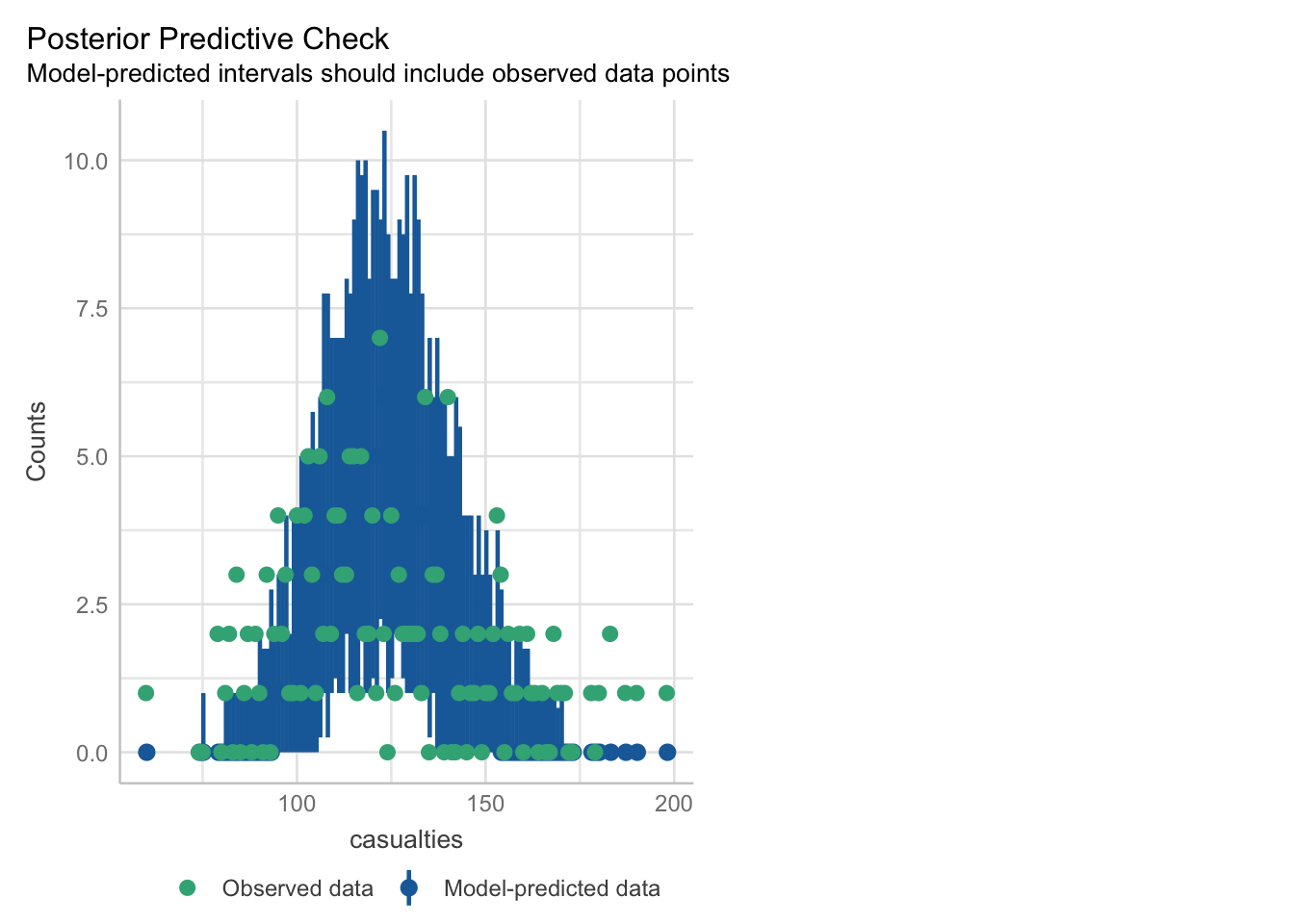
It doesn’t seem to be doing the best job. Around 0 and in the 180+ range of the x-axis, it’s under-predicting the counts; and it might be over-predicting a bit in the middle (120-140 ish).
We can assess outliers by printing a list of high leverage points:
influence = glm_isl.get_influence()
cooks = influence.cooks_distance[0]
influential_points = np.where(cooks > 0.5)[0]
print("Influential points:", influential_points)Influential points: []It seems we’re okay on the outlier front.
For dispersion, let’s calculate the parameter and p-value:
pvalue = chi2.sf(glm_stb.deviance, glm_stb.df_resid)
print(glm_stb.deviance/glm_stb.df_resid, pvalue)4.475851158209689 3.1298697149299994e-84Dispersion is definitely a problem here.
| Response distribution & link function | Yes, based on the info given |
| Independence | Yes, based on the info given |
| Influential points | Yes, no data points identified |
| Dispersion | Clear overdispersion |
Given that we have a problem with overdispersion, we probably wouldn’t proceed with significance testing here under normal circumstances. Instead, we’d want to fit a different GLM that can handle this overdispersion.
For completeness, however, since these are course materials rather than normal circumstances - let’s check: is the model significant over the null?
glm_null <- glm(casualties ~ 1,
family = "poisson",
data = seatbelts)
anova(glm_stb, glm_null)Analysis of Deviance Table
Model 1: casualties ~ year
Model 2: casualties ~ 1
Resid. Df Resid. Dev Df Deviance Pr(>Chi)
1 190 850.41
2 191 984.50 -1 -134.08 < 2.2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1model = smf.glm(formula = "casualties ~ 1",
family = sm.families.Poisson(),
data = seatbelts)
glm_stb_null = model.fit()lrstat = -2*(glm_stb_null.llf - glm_stb.llf)
pvalue = chi2.sf(lrstat, glm_stb_null.df_resid - glm_stb.df_resid)
print(lrstat, pvalue)134.0834016068129 5.23879166948147e-31What does the significant p-value here tell us?
Well, given the difference in degrees of freedom (1), the change in residual deviance between our model and the null model is unexpectedly large. So the model is doing something over and above what the null is doing.
Conclusions
Does this significant comparison against the null allow us to draw any conclusions about whether the seatbelt law affects the number of casualties?
No. Don’t let it tempt you.
The model we’ve constructed here is not a good fit and doesn’t meet all of the assumptions. We need to fit a different type of model that can cope with the overdispersion, which we’ll look into next chapter.
12.10 Summary
Key points
- Count data are bounded between 0 and \(\infty\), with integer increases
- This type of data can be modelled with a Poisson regression, using a log link function
- Poisson regression makes all of the same assumptions as logistic regression, plus an assumption about dispersion