Describe how workflow management systems (WfMS) work and their advantages in complex analysis pipelines.
List some of the most popular WfMS used in the field of bioinformatics.
Recognise the main configuration aspects needed to run a Nextflow pipeline: configuration profiles, cache, resumability, samplesheets.
Apply a standard Nextflow pipelines developed by the nf-core community to a set of sequencing data.
5.1 Overview
Bioinformatic analyses always involve multiple steps where data is gathered, cleaned and integrated to give a final set of processed files of interest to the user. These sequences of steps are called a workflow or pipeline. As analyses become more complex, pipelines may include the use of many different software tools, each requiring a specific set of inputs and options to be defined. Furthermore, as we want to chain multiple tools together, the inputs of one tool may be the output of another, which can become challenging to manage.
Although it is possible to code such workflows using shell scripts, these often don’t scale well across different users and compute setups. To overcome these limitations, dedicated workflow/pipeline management software packages have been developed to help standardise pipelines and make it easier for the user to process their data. These dedicated packages are designed to streamline and automate the process of coordinating complex sequences of tasks and data processing (for instance an RNA-seq analysis). In this way, researchers can focus on their scientific questions instead of the nitty-gritty of data processing.
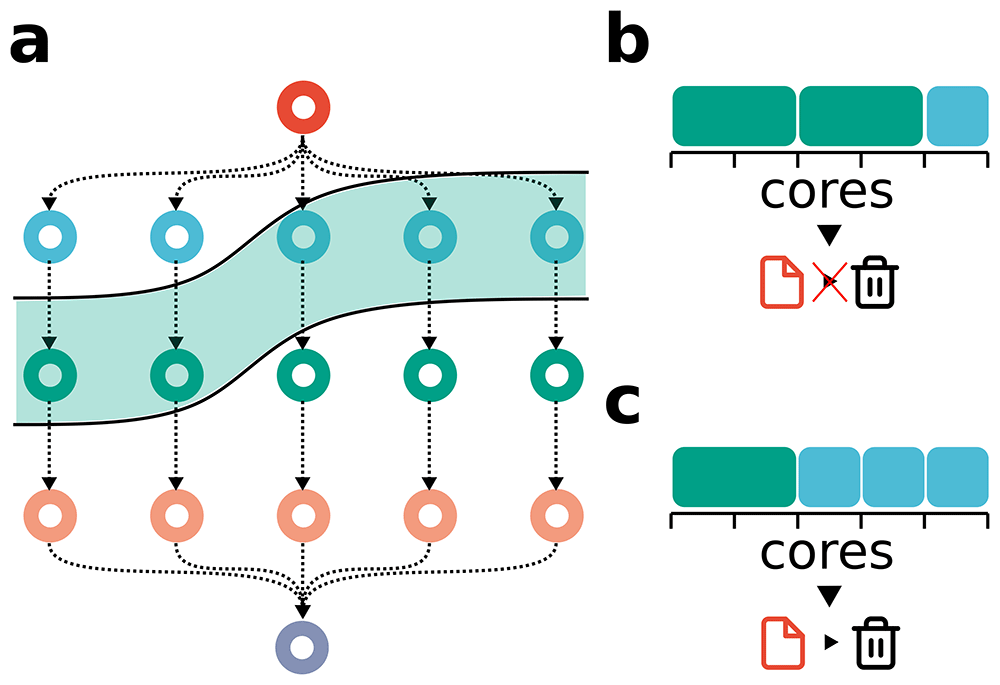
A) Workflow illustration, showing ready-for-running tasks highlighted in green, indicating all necessary input files are available. The initial red task produces a temporary file, potentially removable once the blue tasks complete. Workflow management systems ensure tasks are run in an optimal and automated manner. For example, in B) there is suboptimal scheduling of tasks, as only one blue task is scheduled the temporary file cannot be removed. Conversely, in C) we have optimal scheduling, since the three blue tasks are scheduled first enabling deletion of the temporary file after their completion. Diagram taken from Mölder et al. 2025, licensed under CC BY 4.0).
Here are some of the key advantages of using a standardised workflow for our analysis:
Fewer errors - because the workflow automates the process of managing input/output files, there are less chances for errors or bugs in the code to occur.
Consistency and reproducibility - analysis ran by different people should result in the same output, regardless of their computational setup.
Software installation - all software dependencies are automatically installed for the user using solutions such as Conda/Mamba, Docker and Singularity.
Scalability - workflows can run on a local desktop or scale up to run on high performance compute (HPC) clusters.
Checkpoint and resume - if a workflow fails in one of the tasks, it can be resumed at a later time.
5.2 Nextflow and Snakemake
Two of the most popular workflow software packages are Snakemake and Nextflow. We will not cover how to develop workflows with these packages, but rather how to use existing workflows developed by the community.1 Both Snakemake and Nextflow offer similar functionality and can work on different computer systems, from personal laptops to large cloud-based platforms, making them very versatile. One of the main noticeable difference to those developing pipelines with these tools is that Snakemake syntax is based on Python, whereas Nextflow is based on Groovy. The choice between one of the other is really down to individual preference.
Another important aspect of these projects are the workflows and modules provided by the community:
nf-core: a community project where scientists contribute ready-to-use, high-quality analysis pipelines. This means you don’t have to start from scratch if someone else has already created a workflow for a similar analysis. It’s all about making data analysis more accessible, standardized, and reproducible across the globe.
Snakemake workflow catalog: a searcheable catalog of workflows developed by the community, with instructions and details on how to use them. Although there is some standardisation of these pipelines, they are not as well curated as the ones from nf-core.
These materials will focus on Nextflow, due to the standarised and ready-to-use pipelines available through nf-core.
NoteHow do I install Nextflow and Snakemake?
You can install both of these packages using Mamba:
You may want to check the latest versions available (here and here), which may be different from the ones in the command above.
5.3 Nextflow command line interface
Nextflow has an array of subcommands for the command line interface to manage and execute pipelines. To see all options you can simply run nextflow -h and a list of available top-level options will appear in your terminal’. here we highlight the three we will be using:
nextflow run [options] [pipeline]: will execute a nextflow pipeline
nextflow log: will print the execution history and log information of a pipeline
nextflow clean: will clean up cache and work directories.
The command nextflow run has some useful options:
-profile: defines which configuration profile(s) to use.
-resume: restarts the pipeline where it last stopped (using cached results).
-work-dir: defines the directory where intermediate result files (cache) are stored.
We detail each of these below.
5.3.1 Configuration profile
There are several ways to configure how our Nextflow workflow runs. All nf-core workflows come with some default profiles that we can choose from:
singularity uses Singularity images for software management. This is the recommended (including on HPC systems).
docker uses Docker images for software management.
mamba uses Mamba for software management. This is not recommended as it is known to be slow and buggy.
To use one of these profiles we use the option -profile followed by the name of the configuration profile we wish to use. For example, -profile singularity will use Singularity to manage and run the software.
Sometimes you may want to use custom profiles, or the pipeline you are using is not from the nf-core community. In that case, you can define your own profile. The easiest may be to look at one of the nf-core configuration files and set your own based on that. For example, to set a profile for Singularity, we create a file with the following:
Let’s say we saved this file as nextflow.config. We can then use this profile by running our pipeline with the options -profile singularity -c nextflow.config. You can also specify more than one configuration file in your command as -c myconfig1 -c myconfig2. If you provide multiple config files, they will be merged so that the settings in the first override the same settings appearing in the second, and so on.
NoteWorkflow configuration
Understanding the config file of a nextflow pipeline can be slightly daunting at first, especially if you start with a nf-core configuration. For example, if you look at the default config for the nf-core/rnaseq workflow, you can see how many parameters it includes.
The good news is that normally users won’t need to modify most of these, instead you can just modify the parameters of interest and use -c your_config_profile.config when launching your pipeline.
When a Nextflow pipeline runs it creates (by default) a work directory when you execute the pipeline for the first time. The work directory stores a variety of intermediate files used during the pipeline run, called a cache. The storage of these intermediate files is very important, as it allows the pipeline to resume from a previous state, in case it ran with errors and failed half-way through (more on this below).
Each task from the pipeline (e.g. a bash command can be considered a task) will have a unique directory name within work. When a task is created, Nextflow stages the task input files, script, and other helper files into the task directory. The task writes any output files to this directory during its execution, and Nextflow uses these output files for downstream tasks and/or publishing. Publishing is when the output of a task is being saved to the output directory specified by the user.
The -work-dir option can be used to change the name of the cache directory from the default work. This default directory name is fine (and most people just use that), but you may sometimes want to define a different one. For example, if you coincidentally already have a directory called “work” in your project, or if you want to use a separate storage partition to save the intermediate files.
Regardless, it is important to remember that your final results are not stored in the work directory. They are saved to the output directory you define when you run the pipeline. Therefore, after successfully finishing your pipeline you can safely remove the work directory. This is important to save disk space and you should make sure to do it regularly.
5.3.3 Checkpoint-and-resume
Because Nextflow is keeping track of all the intermediate files it generates, it can re-run the pipeline from a previous step, if it failed half-way through. This is an extremely useful feature of workflow management systems and it can save a lot of compute time, in case a pipeline failed.
All you have to do is use the option -resume when launching the pipeline and it will always resume where it left off. Note that, if you remove the work cache directory detailed above, then the pipeline will have to start from the beginning, as it doesn’t have any intermediate files saved to resume from.
Most nf-core pipelines use a CSV file as their main input file. These CSV file is often referred to as the “sampleshet”, as it contains information about each sample to be processed.
Although there is no universal format for this CSV file, most pipelines accept at least 3 columns:
sample: a name for the sample, which can be anything of our choice.
fastq_1: the path to the respective read 1 FASTQ file.
fastq_2: the path to the respective read 2 FASTQ file. This value is optional and, if missing it is assumed the sample is from single-end sequencing.
These are only the most basic columns, however many other columns are often accepted, depending on the specific pipeline being used. The details for the input CSV samplesheet are usually given in the “Usage” tab of the documentation. We will see some practical examples in Exercise 1.
As FASTQ files are often named using very long identifiers, it’s a good idea to use some command line tricks to save typing and avoid typos. For example, we can create a first version of our file by listing all read 1 files and saving it into a file:
ls reads/*_1.downsampled.fastq.gz > samplesheet.csv
This will give us a good start to create the samplesheet. We can then open this file in a spreadsheet software such as Excel and create the remaining columns. We can copy-paste the file paths and use the “find and replace” feature to replace “_1” with “_2”. This way we save a lot of time of typing but also reduce the risk of having typos in our file paths.
5.4 Demo nf-core pipeline
To demonstrate the use of standard nf-core pipelines, we will use the aptly named nf-core/demo pipeline. This workflow takes a set of FASTQ files as input, runs them through a simple QC step and outputs processed files as well as a MultiQC quality report.
We have named our samples using informative names of our choice, and indicate the path to the respective FASTQ input files. We can then run our workflow as follows (this command is available from scripts/02-run_nfcore_demo.sh):
nextflow run nf-core/demo \-profile"singularity"-revision"1.2.0"\--input"samplesheet.csv"\--outdir"results/qc"\--fasta"genome/Mus_musculus.GRCm38.dna_sm.chr14.fa.gz"
In this case we used the following generic options:
-profile "singularity" indicates we want to use Singularity to manage the software. Nextflow will automatically download containers for each step of the pipeline.
-revision "1.2.0" means we are running version 1.2.0 of the pipeline. It’s a good idea to define the specific version of the pipeline you run, so you can reproduce the results in the future, in case the pipeline changes. You can see the latest versions available from the workflow documentation page.
--input is the samplesheet CSV for this pipeline, which we prepared beforehand using a spreadsheet program such as Excel.
--outdir is the name of the output directory for our results.
--fasta is the reference genome to be used by the pipeline.
When the pipeline starts running, we are given information about its progress, for example:
executor > local (6)
[d1/4efe7b] NFCORE_DEMO:DEMO:FASTQC (control_rep2) | 4 of 4 ✔
[4b/caf73e] NFCORE_DEMO:DEMO:SEQTK_TRIM (drug_rep2) | 1 of 4
[- ] NFCORE_DEMO:DEMO:MULTIQC -
You will also notice that a new directory called work is created. As mentioned above, this is the cache directory, which stores intermediate files and allows the workflow to resume if it fails half-way through (using the -resume option).
Once the pipeline completes (hopefully successfully), we are given a message:
Many nf-core pipelines allow you to specific the name of an organism (using the --genome option) and will automatically download the reference genome and annotation files for you. However, many of these workflows rely on iGenomes, which is not always up-to-date. Therefore, the use of this option is discouraged, as you may miss the latest annotations or use a version of the genome that is incompatible with the rest of your analysis.
Instead, you can download the latest version of the genome and annotations for your species, from sources such as ENSEMBL (vertebrates and non-vertebrates), GENCODE or UCSC.
Most pipelines then have individual options to use these files as input: --fasta (for the reference genome) and --gff/--gtf (for the transcript annotation).
5.4.1 Cleaning up
If you are happy with the results, you can clean the work cache directory to save space. Before actually removing anything, you can see what the clean command would do using the -dry-run (or -n) option:
nextflow clean -n
This will inform you of what the command would remove. If you’re happy with this, you can go ahead and issue to command to -force (or -f) the removal:
nextflow clean -f
The clean command has several options allowing you finer control over what gets deleted, for example the -before and -after options allow you to clean up cached files before or after the specified date/time.
While nextflow clean works well, by default it still leaves behind some files. Usually these don’t occupy much space, but if you want to completely remove the cached files and hidden log files, you can do this manually:
rm-r .nextflow* work
5.5 Troubleshooting
Inevitably workflows may fail, which could be due to several reasons. For example, an error in our command, a mis-formatted samplesheet, missing input files or sometimes even a bug in the pipeline.
When an error occurs, the nextflow command terminates and an error message is printed on the screen (usually in bright red!). The error messages can be quite long and feel difficult to interpret, but often only a small part of the message is relevant, so read it carefully to see if you can spot what the problem is.
For example, we previously got the following error when running the nf-core/demo pipeline. Can you see what the problem was?
Although this is a long message, the cause of the error itself is at the top where we are told “Process requirement exceeds available memory – req: 36 GB; avail: 23.5 GB”.
This means a step of the pipeline must have requested 36GB by default, but we only had 23.5GB on the computer used to run it. In this case, we could have restricted the memory usage with a custom configuration file, which we will discuss in the next section.
5.6 Exercises
In these exercises, you will explore one (or more, if you have time and interest) of the pipelines from the nf-core community, tailored to different areas of genomic analysis. Start with the version of the exercise that aligns best with your data and interests.
There are two tasks for you to complete: (1) preparing an input samplesheet for the pipeline and (2) writing the nextflow command to launch the pipeline.
Note that all of these datasets are downsampled to be small, so they run quickly. They do not represent best practices in experimental design.
ExerciseExercise 1 - Preparing samplesheet
Most nf-core pipelines require a samplesheet as input, which is essentially a CSV file detailing where to find the sequencing files for each sample. The specific format for the samplesheet is workflow-specific, but always detailed in the respective documentation. Your goal in this exercise is to find what the required format for the samplesheet is and create the CSV file to use in the next exercise.
You can create the CSV using a spreadsheet software (such as Excel or LibreOffice Calc).
As a bonus, you can try to create the samplesheet in a less manual way using the provided metadata files. You could do this using command line tools (such as awk or perl) or even using R or Python.
Following the documentation for the pipeline, we could have created our samplesheet in Excel like this (note: the filenames are truncated):
A
B
C
D
1
sample
fastq_1
fastq_2
strandedness
2
inf33_rep1
reads/SRR7657874_1.down …
reads/SRR7657874_2.down …
auto
3
inf33_rep3
reads/SRR7657872_1.down …
reads/SRR7657872_2.down …
auto
4
un11_rep1
reads/SRR7657877_1.down …
reads/SRR7657877_2.down …
auto
5
un11_rep2
reads/SRR7657876_1.down …
reads/SRR7657876_2.down …
auto
However, for many samples, it can be tedious and error-prone to do this kind of task manually. We can see that the FASTQ file names are essentially the prefix given in the metadata file, along with suffixes “_1.downsampled.fastq.gz” and “_2.downsampled.fastq.gz” for read 1 and read 2, respectively.
Therefore, we can create the samplesheet based on our metadata TSV file using some programming tools. Here’s 4 different ways of doing it:
import pandas as pd# read metadata table, ensuring NA values are correctly importedmeta = pd.read_csv("sample_info.tsv", sep="\t", na_values="")# create data frame for output samplesheetout = pd.DataFrame({'sample': meta['name'],'fastq_1': "reads/"+ meta['fastq'] +"_1.downsampled.fastq.gz",'fastq_2': "reads/"+ meta['fastq'] +"_2.downsampled.fastq.gz",'strandedness': "auto"})# save the samplesheet ensuring NA values are written as empty cellsout.to_csv("samplesheet.csv", index=False, quoting=3, na_rep="")
# read metadata table, ensuring NA values are correctly importedmeta <-read.table("sample_info.tsv", header =TRUE, sep ="\t", na.strings ="")# create data frame for output samplesheetout <-data.frame(sample = meta$name, fastq_1 =paste0("reads/", meta$fastq, "_1.downsampled.fastq.gz"), fastq_2 =paste0("reads/", meta$fastq, "_2.downsampled.fastq.gz"),strandedness ="auto")# save the samplesheet ensuring NA values are written as empty cellswrite.csv(out, "samplesheet.csv", row.names =FALSE, quote =FALSE, na ="")
# create new file with the required column namesecho"sample,fastq_1,fastq_2,strandedness"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |perl-ne'chomp;@a=split/\t/;print "$a[4],reads/$a[0]\_1.downsampled.fastq.gz,reads/$a[0]\_2.downsampled.fastq.gz,auto\n"'>> samplesheet.csv
# create new file with the required column namesecho"sample,fastq_1,fastq_2,strandedness"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |awk'BEGIN { FS="\t"; OFS="," }{ print $5, "reads/" $1 "_1.downsampled.fastq.gz", "reads/" $1 "_2.downsampled.fastq.gz", "auto"}'>> samplesheet.csv
Note, if all of the coding suggestions above seem unclear, we reiterate that you can create the samplesheet by hand in a standard spreadsheet software. At the end, our samplesheet should look like this:
Following the documentation for the pipeline, we could have created our samplesheet in Excel like this:
A
B
C
D
E
F
G
1
sample
fastq_1
fastq_2
replicate
antibody
control
control_replicate
2
brd4_veh
reads/SRR1193526.fastq.gz
1
BRD4
mcf7_input_veh
1
3
brd4_veh
reads/SRR1193527.fastq.gz
2
BRD4
mcf7_input_veh
1
4
brd4_e2
reads/SRR1193529.fastq.gz
1
BRD4
mcf7_input_e2
1
5
brd4_e2
reads/SRR1193530.fastq.gz
2
BRD4
mcf7_input_e2
1
6
mcf7_input_veh
reads/SRR1193562.fastq.gz
1
7
mcf7_input_e2
reads/SRR1193563.fastq.gz
1
However, for many samples, it can be tedious and error-prone to do this kind of task manually. We can see that the FASTQ file names are essentially the prefix given in the metadata file, along with the suffix “.fastq.gz” (these are single-end sequencing data, so there’s only one file per sample).
Therefore, we can create the samplesheet based on our metadata TSV file using some programming tools. Here’s 4 different ways of doing it:
import pandas as pd# read metadata table, ensuring NA values are correctly importedmeta = pd.read_csv("sample_info.tsv", sep="\t", na_values="")# create data frame for output samplesheet# "Int64" is used to ensure replicate columns output as integersout = pd.DataFrame({"sample": meta["name"],"fastq_1": "reads/"+ meta["fastq"] +".fastq.gz","fastq_2": pd.NA,"replicate": meta["rep"].astype("Int64"),"antibody": meta["antibody"],"control": meta["input_control"],"control_replicate": meta["input_rep"].astype("Int64")})# save the samplesheet ensuring NA values are written as empty cellsout.to_csv("samplesheet.csv", index=False, quoting=3, na_rep="")
# read metadata table, ensuring NA values are correctly importedmeta <-read.table("sample_info.tsv", header =TRUE, sep ="\t", na.strings ="")# create data frame for output samplesheetout <-data.frame(sample = meta$name, fastq_1 =paste0("reads/", meta$fastq, ".fastq.gz"), fastq_2 =NA,replicate = meta$rep,antibody = meta$antibody,control = meta$input_control, control_replicate = meta$input_rep)# save the samplesheet ensuring NA values are written as empty cellswrite.csv(out, "samplesheet.csv", row.names =FALSE, quote =FALSE, na ="")
# create new file with the required column namesecho"sample,fastq_1,fastq_2,replicate,antibody,control,control_replicate"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |perl-ne'chomp;@a=split/\t/;print "$a[0],reads/$a[2].fastq.gz,,$a[1],$a[3],$a[4],$a[5]\n"'>> samplesheet.csv
# create new file with the required column namesecho"sample,fastq_1,fastq_2,replicate,antibody,control,control_replicate"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |awk'BEGIN { FS="\t"; OFS="," }{ print $1, "reads/" $3 ".fastq.gz", "", $2, $4, $5, $6}'>> samplesheet.csv
Note, if all of the coding suggestions above seem unclear, we reiterate that you can create the samplesheet by hand in a standard spreadsheet software. At the end, our samplesheet should look like this:
Following the documentation for the pipeline, we could have created our samplesheet in Excel like this:
A
B
C
1
sample
fastq_1
fastq_2
2
ZA01
SRR17051908_1.fastq.gz
SRR17051908_2.fastq.gz
3
ZA02
SRR17051923_1.fastq.gz
SRR17051923_2.fastq.gz
4
ZA03
SRR17051916_1.fastq.gz
SRR17051916_2.fastq.gz
5
ZA04
SRR17051953_1.fastq.gz
SRR17051953_2.fastq.gz
6
ZA05
SRR17051951_1.fastq.gz
SRR17051951_2.fastq.gz
7
ZA06
SRR17051935_1.fastq.gz
SRR17051935_2.fastq.gz
8
ZA07
SRR17051932_1.fastq.gz
SRR17051932_2.fastq.gz
9
ZA08
SRR17054503_1.fastq.gz
SRR17054503_2.fastq.gz
However, for many samples, it can be tedious and error-prone to do this kind of task manually. We can see that the FASTQ file names are essentially the prefix given in the metadata file, along with the suffix “_1.fastq.gz” and “_2.fastq.gz” for read 1 and read 2, respectively.
Therefore, we can create the samplesheet based on our metadata TSV file using some programming tools. Here’s 4 different ways of doing it:
import pandas as pd# read metadata table, ensuring NA values are correctly importedmeta = pd.read_csv("sample_info.tsv", sep="\t", na_values="")# create data frame for output samplesheetout = pd.DataFrame({'sample': meta['name'],'fastq_1': "reads/"+ meta['fastq'] +"_1.fastq.gz",'fastq_2': "reads/"+ meta['fastq'] +"_2.fastq.gz"})# save the samplesheet ensuring NA values are written as empty cellsout.to_csv("samplesheet.csv", index=False, quoting=3, na_rep="")
# read metadata table, ensuring NA values are correctly importedmeta <-read.table("sample_info.tsv", header =TRUE, sep ="\t", na.strings ="")# create data frame for output samplesheetout <-data.frame(sample = meta$name, fastq_1 =paste0("reads/", meta$fastq, "_1.fastq.gz"), fastq_2 =paste0("reads/", meta$fastq, "_2.fastq.gz"))# save the samplesheet ensuring NA values are written as empty cellswrite.csv(out, "samplesheet.csv", row.names =FALSE, quote =FALSE, na ="")
# create new file with the required column namesecho"sample,fastq_1,fastq_2"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |perl-ne'chomp;@a=split/\t/;print "$a[0],reads/$a[1]\_1.fastq.gz,reads/$a[1]\_2.fastq.gz\n"'>> samplesheet.csv
# create new file with the required column namesecho"sample,fastq_1,fastq_2"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |awk'BEGIN { FS="\t"; OFS="," }{ print $1, "reads/" $2 "_1.fastq.gz", "reads/" $2 "_2.fastq.gz"}'>> samplesheet.csv
Note, if all of the coding suggestions above seem unclear, we reiterate that you can create the samplesheet by hand in a standard spreadsheet software. At the end, our samplesheet should look like this:
Input barcode directories with FASTQ files in fastq_pass/. This is the directory that is created by standard ONT basecalling software such as Guppy or Dorado.
Metadata for each sample is provided in the sample_info.tsv file, which gives you the name of each sample and their respective barcode folder.
AnswerAnswer
Our metadata file contains information about each sample:
Following the documentation for the pipeline, we could have created our samplesheet in Excel like this:
A
B
1
sample
barcode
2
CH01
1
3
CH02
2
4
CH03
3
5
CH04
4
6
CH05
5
7
CH26
26
8
CH27
27
9
CH28
28
10
CH29
29
11
CH30
30
Compared to the other workflows, this one is a relatively simple format, so given our metadata table, it would be easy to create the required samplesheet in Excel. However, for completeness we also show how to do this using the command line to replace tabs for commas using the sed command:
# create new file with the required column namesecho"sample,barcode"> samplesheet.csv# generate file names from metadatatail-n +2 sample_info.tsv |sed's/\t/,/'>> samplesheet.csv
At the end, our samplesheet should look like this:
Now that you have a samplesheet, you should be ready to write the command to run the pipeline. Then, you can launch the pipeline and watch how Nextflow orchestrates the different steps of the analysis.
Given the complexity of some of these pipelines, we give a skeleton of a command to get you started in each case. However, a few things to remember to do in every case:
Add the -r option to specify the version of the workflow to be used (use the latest available on the respective documentation page).
Include the option to use singularity to manage the software.
Check the parameters being used in the respective documentation page.
To make your code editing easier, make sure to include your command in a shell script and then run it using bash. Also, make sure to activate the Nextflow mamba environment (mamba activate nextflow) before you run the script.
Note: some of these pipelines take a long time to run. Once you initiate the pipeline and it seems to be running successfully, you can indicate to the trainers that you have completed the exercise.
Use the samplesheet created in the previous exercise as input.
The reference genome and annotation are in the genome/ directory.
Note that the options to specify these input files require the full path to the file to be specified. In the code skeleton below we show the trick of using the $PWD environment variable to specify the path relative to the current working directory.
We provide a skeleton of what your command should look like in the script scripts/run_rnaseq.sh. Open that script in a text editor (for example, nano or vim) to fix the code and then run the script using bash.
AnswerAnswer
Here is the fixed nextflow command in our script:
nextflow run nf-core/rnaseq \-r"3.26.0"\-profile"singularity"\--input"samplesheet.csv"\--outdir"results/rnaseq"\--gtf"$PWD/genome/Mus_musculus.GRCm38.102.chr14.gtf.gz"\--fasta"$PWD/genome/Mus_musculus.GRCm38.dna_sm.chr14.fa.gz"\--igenomes_ignore\--skip_alignment
We then ran our script with bash scripts/run_rnaseq.sh. While running, we got the progress of the pipeline printed on the screen.
At the end of the pipeline we could see the results in the results/rnaseq folder. For example, we can open the file results/rnaseq/multiqc/star_salmon/multiqc_report.html to look at a quality control report for the pipeline.
Warning
In the code above we’ve used the option --skip_alignment. In real-world analysis we would not recommend doing this by default, as the alignment provides many useful QC metrics for RNA-seq (e.g. gene body coverage, PCR duplication issues, etc.).
Skipping the alignment does result in a much faster run, so it may be justified, for example, if you are processing a very large number of samples and need to save compute resources.
In this case, we used it to save time, as our purpose is to show how to set and run an nf-core pipeline, not to get precise results.
Chromatin immunoprecipitation sequencing analysis using nf-core/chipseq. Go into the chipseq directory for this version of the exercise.
Use the samplesheet created in the previous exercise as input.
The reference genome and annotation are in the genome/ directory.
Note that the options to specify these input files require the full path to the file to be specified. In the code skeleton below we show the trick of using the $PWD environment variable to specify the path relative to the current working directory.
We provide a skeleton of what your command should look like in the script scripts/run_chipseq.sh. Open that script in a text editor (for example, nano or vim) to fix the code and then run the script using bash.
AnswerAnswer
Here is the fixed nextflow command in our script:
nextflow run nf-core/chipseq \-r"2.1.0"\-profile"singularity"\--input"samplesheet.csv"\--outdir"results/chipseq"\--gtf"$PWD/genome/GRCh38.109.chr21.gtf.gz"\--fasta"$PWD/genome/GRCh38.109.chr21.fasta.gz"\--blacklist"$PWD/genome/ENCFF356LFX_exclusion_lists.chr21.bed.gz"\--read_length 100 \--skip_preseq
We then ran our script with bash scripts/run_chipseq.sh. While running, we got the progress of the pipeline printed on the screen.
At the end of the pipeline we could see the results in the results/chipseq folder. For example, we can open the file results/chipseq/multiqc/broadPeak/multiqc_report.html to look at a quality control report for the pipeline.
Analysis of viral genomes using nf-core/viralrecon. Go into the virus_illumina directory for this version of the exercise.
Use the samplesheet created in the previous exercise as input.
The reference genome, gene annotation and primer locations are in the genome/ directory.
Note that the options to specify these input files require the full path to the file to be specified. In the code skeleton below we show the trick of using the $PWD environment variable to specify the path relative to the current working directory.
We provide a skeleton of what your command should look like in the script scripts/run_viralrecon_illumina.sh. Open that script in a text editor (for example, nano or vim) to fix the code and then run the script using bash.
AnswerAnswer
Here is the fixed nextflow command in our script:
nextflow run nf-core/viralrecon \-r"3.0.0"\-profile"singularity"\--input"samplesheet.csv"\--outdir"results/viralrecon"\--platform"illumina"\--protocol"amplicon"\--gff"$PWD/genome/nCoV-2019.annotation.gff.gz"\--fasta"$PWD/genome/nCoV-2019.reference.fasta"\--primer_bed"$PWD/genome/nCoV-2019.V3.primer.bed"\--skip_assembly--skip_freyja\--skip_pangolin--skip_nextclade
We then ran our script with bash scripts/run_viralrecon.sh. While running, we got the progress of the pipeline printed on the screen.
At the end of the pipeline we could see the results in the results/viralrecon folder. For example, we can open the file results/viralrecon/multiqc/multiqc_report.html to look at a quality control report for the pipeline.
Analysis of viral genomes using nf-core/viralrecon. Go into the virus_ont directory for this version of the exercise.
Use the samplesheet created in the previous exercise as input.
The reference genome, gene annotation and primer locations are in the genome/ directory.
Note that the options to specify these input files require the full path to the file to be specified. In the code skeleton below we show the trick of using the $PWD environment variable to specify the path relative to the current working directory.
We provide a skeleton of what your command should look like in the script scripts/run_viralrecon_ont.sh. Open that script in a text editor (for example, nano or vim) to fix the code and then run the script using bash.
nextflow run nf-core/viralrecon \-r"3.0.0"\-profile"singularity"\--input"samplesheet.csv"\--fastq_dir"fastq_pass/"\--outdir"results/viralrecon"\--platform"nanopore"\--protocol"amplicon"\--genome"MN908947.3"\--primer_set"artic"\--primer_set_version"3"\--artic_minion_model"r941_prom_sup_g5014"\--skip_assembly--skip_freyja\--skip_pangolin--skip_nextclade
We then ran our script with bash scripts/run_viralrecon.sh. While running, we got the progress of the pipeline printed on the screen.
At the end of the pipeline we could see the results in the results/viralrecon folder. For example, we can open the file results/viralrecon/multiqc/medaka/multiqc_report.html to look at a quality control report for the pipeline.
5.7 Summary
TipKey points
WfMS define, automate and monitor the execution of a series of tasks in a specific order. They improve efficiency, reduce errors, can be easily scaled (from a local computer to a HPC cluster) and increase reproducibility.
Popular WfMS in bioinformatics include Nextflow and Snakemake. Both of these projects have associated community-maintained workflows, with excellent documentation for their use: nf-core and the snakemake workflow catalog.
Nextflow pipelines have configuration profiles available to indicate how software should be managed by the pipeline. For example the option -profile singularity uses Singularity images to deploy the software (other options include docker and conda).
Nextflow keeps track of the current status of the pipeline by storing intermediate files in a cache directory (by default called “work”). This enables the workflow to resume from a previous run, in case of failure.
Pipelines from the nf-core community commonly take as an input a CSV file detailing the input files for the workflow. This CSV file is commonly referred to as a samplesheet.
Nf-core pipelines have extensive documentation at nf-co.re/pipelines, allowing the user to configure many aspects of the run.

{kind=link}